From 11:00 pm to 12:00 pm EST ( 8:00 pm to 9:00 pm PST ) on January 6th, the website will be under maintenance. We are sorry for the inconvenience. Please arrange your schedule properly.
KG5 is an orally active dual PDGFRβ and B-Raf allosteric inhibitor. KG5 also inhibits Flt3, KIT and c-Raf. KG5 has anticancer, antiangiogenic activities .
C-RAF kinase-IN-1 (compound 1l) is a potent inhibitor of C-RAF kinase with an IC50 of 0.193 μM. C-RAF kinase-IN-1 is a quinoline derivative. C-RAF kinase-IN-1 has the potential for the research of cancer diseases .
Raf inhibitor 3 (Example 30) is a Raf inhibitor. Raf inhibitor 3 inhibits B-Raf and C-Raf with IC50 values less than 15 nM. Raf inhibitor 3 can be used for research of cancers .
BRAF V600E/CRAF-IN-1 (Compound 8b) is a potent inhibitor of BRAF V600E/CRAF. BRAF V600E/CRAF-IN-1 triggers apoptosis and cell cycle arrest at G0/G1 phase in HCT-116 colon cancer cell. BRAF V600E/CRAF-IN-1 has the potential for the research of cancer diseases .
BRAF V600E/CRAF-IN-2 (Compound 9c) is a potent inhibitor of BRAF V600E/CRAF with IC50s of 0.888 and 0.229 μM, respectively. BRAF V600E/CRAF-IN-2 triggers apoptosis and cell cycle arrest at G0/G1 phase in HCT-116 colon cancer cell. BRAF V600E/CRAF-IN-2 has the potential for the research of cancer diseases .
ML786 dihydrochloride is a potent and orally bioavailable Raf inhibitor, with IC50s of 2.1, 4.2, and 2.5 nM for V600EΔB-Raf, wt B-Raf, and C-Raf, respectively. ML786 dihydrochloride also inhibits Abl-1, DDR2, EPHA2, KDR, and RET (IC50=<0.5, 7.0, 11, 6.2, 0.8 nM). ML786 dihydrochloride can be used for the research of cancers .
Kobe0065 is a novel and effective inhibitor of Ras-Raf interaction, competitively inhibiting the binding of H-Ras·GTP to c-Raf-1 RBD with a Ki value of 46±13 μM.
B-Raf inhibitor 1 dihydrochloride is a potent Raf kinase inhibitor with Kis of 1 nM, 1 nM, and 0.3 nM for B-Raf WT, B-Raf V600E, and C-Raf, respectively.
SB-590885 is a potent B-Raf inhibitor with a Ki of 0.16 nM, and has 11-fold greater selectivity for B-Raf over c-Raf, without inhibition to other human kinases.
Kobe2602 is a Ras-Raf interaction inhibitor. Kobe2602 inhibits the binding of H-Ras·GTP to c-Raf-1 RBD with a Ki of 149 μM. Kobe2602 has antitumor activity .
Belvarafenib (HM95573) is a potent and pan RAF (Rapidly Accelerated Fibrosarcoma) inhibitor, with IC50s of 56 nM, 7 nM and 5 nM for B-RAF, B-RAF v600E and C-RAF respectively .
Belvarafenib TFA (HM95573 TFA) is a potent and pan RAF (Rapidly Accelerated Fibrosarcoma) inhibitor, with IC50s of 56 nM, 7 nM and 5 nM for B-RAF, B-RAFv 600E and C-RAF respectively .
(Z)-GW 5074 is a compound which interacts with both mHTT (mutant huntingtin protein) and LC3, but not but not with the wild-type HTT protein. (Z)-GW 5074 inhibits c-Raf, shows no effect on autophagy, and is effective for neurodegenerative disorder .
Dabrafenib-d9 is the deuterium labeled Dabrafenib. Dabrafenib (GSK2118436A) is an ATP-competitive inhibitor of Raf with IC50s of 5 nM and 0.6 nM for C-Raf and B-RafV600E, respectively[4].
Dabrafenib (Standard) is the analytical standard of Dabrafenib. This product is intended for research and analytical applications. Dabrafenib (GSK2118436A) is an ATP-competitive inhibitor of Raf with IC50s of 5 nM and 0.6 nM for C-Raf and B-Raf V600E, respectively .
Vemurafenib (PLX4032) is a first-in-class, selective, potent inhibitor of B-RAF kinase, with IC50s of 31 and 48 nM for RAF V600E and c-RAF-1, respectively . Vemurafenib induces cell autophagy .
GW 5074 is a potent and selective c-Raf inhibitor with IC50 of 9 nM, and has no effect on the activities of JNK1/2/3, MEK1, MKK6/7, CDK1/2, c-Src, p38 MAP, VEGFR2 or c-Fms .
RAF mutant-IN-1 is a RAF kinase inhibitor, extracted from patent WO2019107987A1, with IC50 values of 21 nM, 30 nM and 392 nM for C-RAF 340D/Y341D, B-RAF V600E and B-RAF WT, respectively .
Vemurafenib-d7 is deuterium labeled Vemurafenib. Vemurafenib (PLX4032) is a first-in-class, selective, potent inhibitor of B-RAF kinase, with IC50s of 31 and 48 nM for RAFV600E and c-RAF-1, respectively[1][4]. Vemurafenib induces cell autophagy[5].
Vemurafenib-d5 is the deuterium labeled Vemurafenib. Vemurafenib (PLX4032) is a first-in-class, selective, potent inhibitor of B-RAF kinase, with IC50s of 31 and 48 nM for RAFV600E and c-RAF-1, respectively[1][4]. Vemurafenib induces cell autophagy[5].
SB-590885 (Standard) is the analytical standard of SB-590885 (HY-10966). This product is intended for research and analytical applications. SB-590885 is a potent B-Raf inhibitor with a Ki of 0.16 nM, and has 11-fold greater selectivity for B-Raf over c-Raf, without inhibition to other human kinases.
Vemurafenib (Standard) is the analytical standard of Vemurafenib. This product is intended for research and analytical applications. Vemurafenib (PLX4032) is a first-in-class, selective, potent inhibitor of B-RAF kinase, with IC50s of 31 and 48 nM for RAF V600E and c-RAF-1, respectively . Vemurafenib induces cell autophagy .
AFG210 is a potent multi-target kinase inhibitor that primarily inhibits Abl kinase (IC50=330 nM), and also has inhibitory effects on other kinases such as B-Raf, C-Raf, FGFR-1, RET and VEGF receptors. AFG210 can be used to study chronic myeloid leukemia and other diseases with abnormal activation of Abl kinase .
PLX-4720 is a potent and selective inhibitor of B-Raf V600E with IC50 of 13 nM in a cell-free assay, equally potent to c-Raf-1(Y340D and Y341D mutations), and 10-fold selectivity for B-Raf V600E than wild-type B-Raf.
OSI-930 is an orally selective inhibitor of Kit, KDR and CSF-1R (c-Fms) with IC50s of 80 nM, 9 nM and 15 nM, respectively. OSI-930 also moderately inhibits Flt-1, c-Raf, Lck and low activity against PDGFRα/β, Flt-3 and Abl. OSI-930 has antitumor activity .
ML786 is a potent and orally bioavailable Raf inhibitor, with IC50s of 2.1, 4.2, and 2.5 nM for V600EΔB-Raf, wt B-Raf, and C-Raf, respectively. ML786 also inhibits Abl-1, DDR2, EPHA2, KDR, and RET (IC50=<0.5, 7.0, 11, 6.2, 0.8 nM). ML786 can be used for the research of cancers .
PLX-4720-d7 is the deuterium labeled PLX-4720. PLX-4720 is a potent and selective inhibitor of B-RafV600E with an IC50 of 13 nM in a cell-free assay, equally potent to c-Raf-1(Y340D and Y341D mutations), and 10-fold selectivity for B-RafV600E than wild-type B-Raf[1][2].
AS601245 is an orally active, selective, ATP competitive JNK (c-Jun NH2-terminal protein kinase) inhibitor with IC50s of 150, 220, and 70 nM for three JNK human isoforms (hJNK1, hJNK2, and hJNK3), respectively. AS601245 exhibits 10- to 20-fold selectivity over c-src, CDK2, and c-Raf and more than 50- to 100-fold selectivity over a range of Ser/Thr- and Tyr-protein kinases. Neuroprotective properties .
AS601245 TFA is an orally active, selective, ATP competitive JNK (c-Jun NH2-terminal protein kinase) inhibitor with IC50s of 150, 220, and 70 nM for three JNK human isoforms (hJNK1, hJNK2, and hJNK3), respectively. AS601245 TFA exhibits 10- to 20-fold selectivity over c-src, CDK2, and c-Raf and more than 50- to 100-fold selectivity over a range of Ser/Thr- and Tyr-protein kinases. Neuroprotective properties .
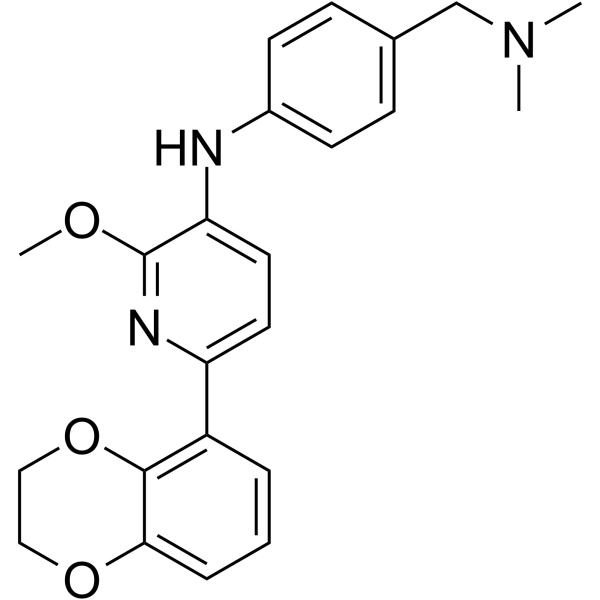
Naporafenib (LXH254) is a potent, selective, orally active, type II BRAF and CRAF inhibitor, with IC50 values of 0.072 and 0.21 nM against CRAF and BRAF, respectively .
SKLB646 is an orally active multi-target kinase inhibitor. SKLB646 shows significant inhibitory effects on SRC and VEGFR2 with IC50 values ??of 0.002 μmol/L and 0.012 μmol/L, respectively. SKLB646 also shows significant inhibitory effects on B-Raf and C-Raf with IC50 values ??of 0.022 μmol/L and 0.019 μmol/L, respectively. SKLB646 inhibits the activation of the SRC signaling pathway and blocks the MAPK signaling pathway by inhibiting Raf kinase. In addition, SKLB646 can inhibit the proliferation, migration and invasion of human umbilical vein endothelial cells (HUVEC) to inhibit tumor-induced angiopoietic formation. SKLB646 shows significant anti-proliferative and anti-survival activities against triple-negative breast cancer (TNBC) cell lines .
RAF-IN-1 is a potent b/cRAF inhibitor with an IC50s of 3.8 nM, 36 nM, 29.4 nM for cRAF, bRAF wt, and bRAF V600E. RAF-IN-1 shows cell growth inhibition with GI50s of 3.4 and 2.9 nM for H358 and A375 cell line bearing bRAF V600E mutation, respectively .
PF-04880594 is a potent and selective RAF inhibitor. PF-04880594 inhibits both wild-type and mutant BRAF and CRAF. PF-04880594 shows antitumor activity .
XL-281 (BMS-908662) is an orally active inhibitor for RAF kinase, with IC50s of 2.6, 4.5 and 6 nM, for CRAF, B-RAF, and B-RAFV600E, respectively. XL-281 exhibits antitumor activity .
CCT241161 is an orally active pan-RAF inhibitor with IC50s of 3, 6, 10, 15 and 30 nM for LCK, CRAF, SRC, V600E-BRAF and BRAF, respectively. CCT241161 shows good activity to in BRAF and NRAS mutant melanomas. CCT241161 also exhibits anticancer cell proliferative activity .
AZ304 is an ATP-competitive dual BRAF kinase inhibitor, potently inhibits wild type BRAF, V600E mutant BRAF and wild type CRAF, with IC50s of 79 nM, 38 nM and 68 nM, respectively. AZ304 also has significant effect on other kinases, such as p38 (IC50, 6 nM), CSF1R (IC50, 35 nM). Anti-tumor activity .
Pan-Raf/RTK inhibitor 1 (compound I-16) is a potent pan-Raf inhibitor with IC50 values of 3.49 nM (BRaf V600E), 8.86 nM (ARaf), 5.78 nM (BRaf WT), and 1.65 nM (CRaf). Pan-Raf/RTK inhibitor 1 exhibits antiproliferative activities against various cancer cell lines and can be utilized in cancer research .
TH-Z827 is a mutant selective KRAS(G12D) inhibitor with an IC50 of 2.4 μM. TH-Z827 does not bind KRAS(WT) or KRAS(G12C). TH-Z827 blocked the KRAS(G12D)-CRAF interaction with an IC50 value of 42 μM .
GNE-9815 (compound 7) is a highly selective, pan-RAF inhibitor with good oral bioavailability. GNE-9815 exhibits Ki values of 0.062 and 0.19 nM for CRAF and BRAF, respectively. GNE-9815 combines with MEK inhibitor Cobimetinib (HY-13064) shows synergistic modulation of MAPK pathway. GNE-9815 can be used in studies of KRAS mutant cancers .
BI-882370 is a potent and selective RAF kinase inhibitor that binds to the ATP binding site of the kinase positioned in the DFG-out (inactive) conformation of the BRAF kinase. BI-882370 (BI 882370) inhibits the oncogenic BRAF V600E-mutant, the WT BRAF and CRAF kinases with IC50s of 0.4, 0.8, and 0.6 nM, respectively. BI-882370 also inhibits SRC family kinases .
NST-628 is a brain-permeable MAPK pathway molecule glue that inhibits RAF phosphorylation and MEK activation. NST-628 also binds RAF and prevents the formation of BRAF-CRAF and BRAF-ARAF heterodimers, effectively inhibiting the RAS-MAPK pathway. NST-628 inhibits RAS- and RAF-driven cancers and demonstrated potent inhibition in mutant KRAS, NRAS, BRAF class II/III, and NF1-mutant tumors .
SHR902275 is a potent, selective, and orally active RAF inhibitor targeting RAS mutant cancers. SHR902275 has IC50s of 1.6 nM, 10 nM, and 5.7 nM for cRAF, bRAF wt, and bRAF V600E, respectively. SHR902275 shows cell growth inhibition with GI50s of 1.5 and 0.17 nM, 0.4 nM and 0.32 nM for H358, A375, Calu6, and SK-MEL2 cells respectively .
MS934 is a novel improved VHL-recruiting MEK 1/2 PROTAC degrader. MS934 also degrades CRAF. MS934 can be used for the research of variety of human cancers, such as melanoma, nonsmall cell lung cancer (NSCLC), colorectal cancer, primary brain tumors, and hepatocellular carcinoma (Pink: Target protein ligand (HY-168288); Black: linker (HY-168289); Blue: E3 ligase ligand (HY-112078)) .
RAS inhibitor Abd-7, a potent RAS-binding compound (Kd=51 nM), is a RAS-effector protein-protein interaction (PPI) inhibitor. RAS inhibitor Abd-7 interacts with RAS inside the cells, prevents RAS-effector interactions and inhibits endogenous RAS-dependent signaling. RAS inhibitor Abd-7 impairs the PPI of various mutant KRAS proteins with PI3K, CRAF and RALGDS as well as NRAS Q61H and HRAS G12V .
Antitumor agent-60 (compound 20) is a potent antitumor agent, targeting RAS-RAF signaling pathway and binding to CRAF with a Kd value of 3.93 μM. Antitumor agent-60 induces apoptosis by blocking cell cycle at G2/M phase. Antitumor agent-60 enhances the level of p53 and ROS. Antitumor agent-60 causes oval and irregular nucleus in cancer cells. Antitumor agent-60 can suppress the growth of tumor to some extent in A549 xenograft model .
Avutometinib (CH5126766) (potassium) is a RAF/MEK clamp that potently inhibits RAF/MEK kinase activity and induces dominant negative RAF-MEK complexes preventing phosphorylation of MEK by ARAF, BRAF and CRAF. Avutometinib (potassium) shows anti-proliferative potency across tumor cell lines carrying KRAS mutations including PDAC cell lines. Avutometinib (potassium) induces tumor inhibition and increases survival in a KRAS/p53 pancreatic cancer mouse model. Avutometinib (potassium) is promising for research of low-grade-serous-ovarian-carcinoma (LGSOC), ovarian cancer and pancreatic ductal adenocarcinoma (PDAC) .
RMC-7977 is an orally active triple-complex RAS inhibitor that can simultaneously bind to cyclophilin A (CYPA) (Kd = 195 nM) and KRAS (G12V) (Kd = 292 μM). It exhibits broad-spectrum inhibitory activity against KRAS, NRAS, and HRAS proteins and their various wild-type and mutant variants. RMC-7977 induces apoptosis by inhibiting the phosphorylation of ERK, CRAF, and RSK, as well as increasing PARP cleavage. This leads to tumor regression, reduces resistance in KRAS G12C cancer models, and demonstrates good tolerability across various RAS cancer models .
SJ-C1044 is an orally available pan-RAF inhibitor with immunomodulatory and anti-tumor activities. SJ-C1044 inhibits wild-type BRAF, wild-type CRAF, and BRAF (V600E) with IC50 values ??of 331, 257, and 187 nM, respectively. SJ-C1044 inhibits tumor cell proliferation by inhibiting kras activation and MEK-ERK phosphorylation. In addition, SJ-C1044 also has a certain inhibitory effect on VEGFR2, TIE2, and CSF1R, with IC50 values ??of 100, 23, and 235 nM, respectively. SJ-C1044 improves the tumor immune microenvironment by inhibiting angiogenesis and regulating macrophage function. SJ-C1044 can be used in the study of colorectal cancer .
RAF1 is a kinase that links Ras GTPases to the MAPK/ERK cascade, influencing cell fate decisions. Activated RAF1 initiates the MAPK cascade and phosphorylates MEK1/2 and ERK1/2. RAF1 Protein, Human (Y340D,Y341D, sf9, His, GST) is the recombinant human-derived RAF1, expressed by Sf9 insect cells , with His, GST labeled tag. ,
Dabrafenib-d9 is the deuterium labeled Dabrafenib. Dabrafenib (GSK2118436A) is an ATP-competitive inhibitor of Raf with IC50s of 5 nM and 0.6 nM for C-Raf and B-RafV600E, respectively[4].
Vemurafenib-d7 is deuterium labeled Vemurafenib. Vemurafenib (PLX4032) is a first-in-class, selective, potent inhibitor of B-RAF kinase, with IC50s of 31 and 48 nM for RAFV600E and c-RAF-1, respectively[1][4]. Vemurafenib induces cell autophagy[5].
Vemurafenib-d5 is the deuterium labeled Vemurafenib. Vemurafenib (PLX4032) is a first-in-class, selective, potent inhibitor of B-RAF kinase, with IC50s of 31 and 48 nM for RAFV600E and c-RAF-1, respectively[1][4]. Vemurafenib induces cell autophagy[5].
PLX-4720-d7 is the deuterium labeled PLX-4720. PLX-4720 is a potent and selective inhibitor of B-RafV600E with an IC50 of 13 nM in a cell-free assay, equally potent to c-Raf-1(Y340D and Y341D mutations), and 10-fold selectivity for B-RafV600E than wild-type B-Raf[1][2].
Raf1 Antibody is a non-conjugated and Rabbit origined monoclonal antibody about 73 kDa, targeting to Raf1. It can be used for WB,ICC/IF assays with tag free, in the background of Human, Mouse, Rat.