Search Result
Results for "
mutant selective
" in MedChemExpress (MCE) Product Catalog:
8
Isotope-Labeled Compounds
| Cat. No. |
Product Name |
Target |
Research Areas |
Chemical Structure |
-
- HY-19706
-
ARS-853
4 Publications Verification
|
Ras
Apoptosis
|
Cancer
|
|
ARS-853 is a cell-active, selective, covalent KRAS G12C inhibitor with an IC50 of 2.5 μM. ARS-853 inhibits mutant KRAS-driven signaling by binding to the GDP-bound oncoprotein and preventing activation .
|
-

-
- HY-10624
-
THIQ
1 Publications Verification
|
Melanocortin Receptor
|
Metabolic Disease
|
|
THIQ is the first selective agonist of the melanocortin-4 receptor (MC4R), with high affinity and potency for hMC4R (IC50=1.2 nM, EC50=2.1 nM) and rMC4R (IC50=0.6 nM, EC50=2.9 nM). THIQ maintains low potency at MC1R, MC3R and MC5R. THIQ plays a role in eliciting erectile activity in rodents. THIQ acts as a pharmacoperone of the MC4R rescuing the cell surface expression and signaling of some intracellularly retained MC4R mutants .
|
-

-
- HY-157229
-
|
|
EGFR
|
Others
|
|
STX-721 is a mutant-selective inhibitor of EGFR and HER2 Exon 20 insertion (ex20ins) mutants .
|
-

-
- HY-19803
-
|
ASP8273 mesylate
|
EGFR
|
Cancer
|
|
Naquotinib mesylate (ASP8273 mesylate) is an orally available, mutant-selective and irreversible EGFR inhibitor; with IC50s of 8-33 nM toward EGFR mutants and 230 nM for EGFR.
|
-

-
- HY-19729
-
|
ASP8273
|
EGFR
|
Cancer
|
|
Naquotinib (ASP8273) is an orally available, mutant-selective and irreversible EGFR inhibitor; with IC50s of 8-33 nM toward EGFR mutants and 230 nM for EGFR.
|
-

-
- HY-19985
-
|
|
EGFR
|
Cancer
|
|
PF-06459988 is an orally activity, irreversible and mutant-selective inhibitor of EGFR mutant forms. PF-06459988 demonstrates high potency and specificity to the T790M-containing double mutant EGFRs. PF-06459988 can be used for the research of cancer .
|
-

-
- HY-12872
-
|
EGF816
|
EGFR
|
Cancer
|
|
Nazartinib (EGF816) is a covalent mutant-selective EGFR inhibitor, with Ki and Kinact of 31 nM and 0.222 min −1 on EGFR(L858R/790M) mutant, respectively.
|
-

-
- HY-P3277
-
|
|
Ras
|
Cancer
|
|
KRpep-2d is a potent K-Ras inhibitor and inhibits proliferation of K-Ras-driven cancer cells. KRpep-2d can be used for cancer research .
|
-

-
- HY-100476
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Mutant IDH1-IN-3 (Compound 1) is a selective allosteric inhibitor of mutant isocitrate dehydrogenase 1 (IDH1). Mutant IDH1-IN-3 has an IC50 of 13 nM for R132H IDH1. Mutant IDH1-IN-3 inhibits the production of D-2-hydroxyglutaric acid (2HG) in cells. Mutant IDH1-IN-3 can be used in the study of cancer .
|
-

-
- HY-100520
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-2 is a a noncovalent, irreversible, mutant-selective second generation EGFR inhibitor.
|
-

-
- HY-P3277A
-
|
|
Ras
|
Cancer
|
|
KRpep-2d (TFA) is a potent K-Ras inhibitor and inhibits proliferation of K-Ras-driven cancer cells. KRpep-2d can be used for cancer research .
|
-

-
- HY-12872A
-
|
EGF816 mesylate
|
EGFR
|
Cancer
|
|
Nazartinib mesylate (EGF816 mesylate) is a novel, covalent mutant-selective EGFR inhibitor, with Ki and Kinact of 31 nM and 0.222 min ?1 on EGFR(L858R/790M) mutant, respectively.
|
-

-
- HY-18690A
-
-

-
- HY-13984
-
|
|
EGFR
|
Cancer
|
|
Mutant EGFR inhibitor is a potent and selective mutant EGFR inhibitor extracted from patent WO 2013014448 A1; inhibits EGFR L858R, EGFR Exon 19 deletion and EGFR T790M.
|
-
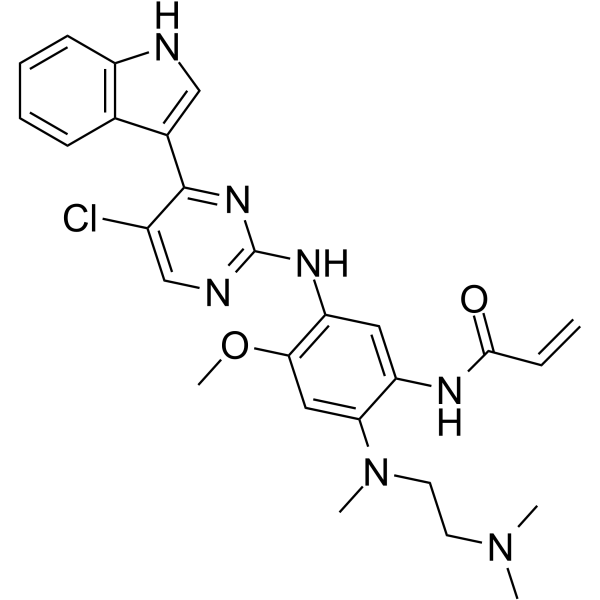
-
- HY-13897
-
|
|
EGFR
|
Cancer
|
|
CNX-2006 is a mutant-selective and irreversible EGFR inhibitor with an IC50 below 20 nM for EGFR T790M.
|
-

-
- HY-12408
-
|
|
Ras
|
Cancer
|
|
6H05 is a selective, and allosteric inhibitor of oncogenic mutant K-Ras(G12C).
|
-

-
- HY-160287
-
|
|
Epigenetic Reader Domain
|
Cancer
|
|
NVS MLLT-1 (compound 3) is a potent and selective MLLT1 inhibitor with Kd values of 0.18 µM, 0.24 µM, and 0.22 µM for wild-type, T1 mutants, and T3 mutants, respectively .
|
-

-
- HY-W423595
-
|
|
EGFR
|
Cancer
|
|
BEBT-109 is a potent pan-mutant-selective EGFR inhibitor. BEBT-109 has improved pharmacokinetic properties. BEBT-109 can be used for multiple mutant-EGFR-driven non-small cell lung cancer (NSCLC) research .
|
-

-
- HY-131312
-
|
LY3410738; mutant IDH1-IN-6
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Mutant IDH1-IN-6 is a potent, selective and orally active mutant isocitrate dehydrogenase (IDH) inhibitor with IC50s of 6.27 nM, 3.71 nM, 36.9 nM and 11.5 nM for IDH1 R132H, IDH1 R132C, IDH2 R140Q and IDH2 R172K mutant enzymes, respectively. Mutant IDH1-IN-6 is less active at inhibiting the IDH wild-type enzymes .
|
-

-
- HY-119177
-
|
|
Bcr-Abl
|
Others
|
|
PD173956 is a pyridopyrimidine Bcr-Abl kinase inhibitor with high selectivity for the P-loop mutant form of Bcr-Abl .
|
-

-
- HY-12408A
-
|
|
Ras
|
Cancer
|
|
6H05 TFA is a selective, and allosteric inhibitor of oncogenic mutant K-Ras(G12C).
|
-

-
- HY-149359
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
IHMT-IDH1-053 (compound 16) is a highly selectivity and irreversible IDH1-mutant inhibitor with an IC50 of 4.7 nM for IDH1 R132H. IHMT-IDH1-053 displays high selectivity against IDH1 mutants over IDH1 wt and IDH2 wt/mutants. IHMT-IDH1-053 inhibits 2-hydroxyglutarate (2-HG) production in IDH1 R132H mutant transfected 293T cells (IC50=28 nM). IHMT-IDH1-053 binds to the IDH1 R132H protein in the allosteric pocket adjacent to the NAPDH binding pocket through a covalent bond with residue Cys269. IHMT-IDH1-053 inhibits the proliferation of HT1080 cell line and primary AML cells which both bear IDH1 R132 mutants .
|
-

-
- HY-156681A
-
|
|
PI3K
|
Cancer
|
|
(S)-STX-478 is the S-enantiomer of STX-478. STX-478 is a selective inhibitor of PI3Kα mutants, preventing metabolic dysfunction and demonstrating antitumor activity in xenograft mouse models with PI3Kα mutant tumors .
|
-

-
- HY-18997
-
PLX7904
3 Publications Verification
PB04
|
Raf
|
Cancer
|
|
PLX7904 is a potent and selective BRAF inhibitor, with IC50 of appr 5 nM against BRAF V600E in mutant RAS expressing cells.
|
-

-
- HY-15003
-
|
|
FLT3
Apoptosis
|
Cancer
|
|
ATH686 is a potent, selective and ATP-competitive FLT3 inhibitor. ATH686 target mutant FLT3 protein kinase activity and inhibit the proliferation of cells harboring FLT3 mutants via induction of apoptosis and cell cycle inhibition. ATH686 has antileukemic effects .
|
-

-
- HY-153306
-
|
|
PI3K
|
Cancer
|
|
RLY-2608 is an orally active first-in-class allosteric mutant-selective inhibitor of PI3Ka with anti-tumor activity. RLY-2608 inhibits tumor growth in PIK3CA-mutant xenograft mice models with minimal impact on insulin .
|
-

-
- HY-12475
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Mutant IDH1-IN-1 is a mutant-selective IDH1 inhibitor with with IC50s of 4, 42, 80 and 143 nM against mutant IDH1 R132C/R132C, IDH1 R132H/R132H, IDH1 R132H/WT and wild type IDH1, respectively.
|
-

-
- HY-18082
-
-

-
- HY-104036
-
IDH-305
1 Publications Verification
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
IDH-305 is an orally available, mutant-selective and brain-penetrant IDH1 inhibitor that targets IDH1 (R132) mutation. IDH-305 exhibits greater than 200 fold selectivity for mutant IDH1 isoforms vs. WT (IC50= 27 nM (IDH1 R132H), 28 nM (IDH1 R132C), 6.14 µM (IDH1 WT)) .
|
-

-
- HY-154959
-
|
|
Ras
|
Cancer
|
|
AZD4747 is a selective, blood-brain barrier-permeable mutant GTPase KRAS G12C inhibitor. AZD4747 has the potential to study cancer .
|
-

-
- HY-111050
-
|
|
c-Met/HGFR
|
Cancer
|
|
JNJ-38877618 is a potent, highly selective, orally bioavailable Met kinase inhibitor with IC50s of 2 and 3 nM for wild type and mutant Met, respectively.
|
-

-
- HY-125841
-
|
|
EGFR
|
Cancer
|
|
EGFR mutant-IN-1, a 5-methylpyrimidopyridone derivative, is a potent and selective EGFR L858R/T790M/C797S mutant inhibitor with an IC50 of 27.5 nM, while being a significantly less potent for EGFR WT (IC50 >1.0 μM) .
|
-

-
- HY-128860
-
|
|
EGFR
|
Cancer
|
|
Mutated EGFR-IN-2 (compound 91) is a mutant-selective EGFR inhibitor extracted from patent WO2017036263A1, which potently inhibits single-mutant EGFR (T790M) and double-mutant EGFR (including L858R/T790M (IC50=<1nM) and ex19del/T790M), and can suppress activity of single gain-of-function mutant EGFR (including L858R and ex19del) as well. Mutated EGFR-IN-2 shows anti-tumor antivity .
|
-

-
- HY-128589
-
|
|
c-Kit
|
Cancer
|
|
CHMFL-KIT-033 is a potent and selective inhibitor of c-KIT T670I mutant for gastrointestinal stromal tumors (GISTs), with an IC50 of 0.045 μM .
|
-

-
- HY-154313
-
|
Clospirazine
|
Ras
|
Cancer
|
|
Spiclomazine (Clospirazine) is a potent mutant KRAS(G12C) inhibitor that selectively inhibits mutant KRAS-driven pancreatic cancer. Spiclomazine can eliminate KRas-GTP levels in KRAS-driven pancreatic cancer and effectively inhibit RAS-mediated signaling. Spiclomazine significantly inhibits tumor progression in mouse renal capsule xenotransplantation models .
|
-

-
- HY-100002
-
-

-
- HY-141807
-
|
|
PROTACs
Akt
|
Cancer
|
|
MS21, a novel degrader of AKT, selectively inhibits the growth of PI3K/PTEN pathway-mutant cancers with wild-type KRAS and BRAF.
|
-

-
- HY-146223
-
|
|
Ras
|
Cancer
|
|
AZD4625 (Compound 21) is a highly potent, selective, covalent and allosteric inhibitor of the mutant GTPase KRAS G12C. AZD4625 has high oral bioavailability .
|
-

-
- HY-117540
-
|
|
Histone Methyltransferase
|
Cancer
|
|
ZLD10A is a highly potent and selective EZH2 inhibitor with the activity of inhibiting H3K27 methylation. ZLD10A can inhibit wild-type and mutant EZH2 with nanomolar potency and has more than 1000-fold selectivity for the other 10 histone methyltransferases. ZLD10A inhibited cell proliferation of DLBCL cell lines in a concentration- and time-dependent manner, showing a potential antiproliferative effect. ZLD10A can be used in the study of EZH2 mutant lymphomas .
|
-

-
- HY-136379
-
|
Cdc42-IN-1
|
Ras
|
Cancer
|
|
CID44216842 (Cdc42-IN-1) is a potent Cdc42-selective guanine nucleotide binding lead inhibitor. The EC50s for Cdc42 WT and Cdc42Q61L mutant are 1.0 and 1.2 μM in GTP binding assay, respectively. The EC50s for Cdc42 WT and Cdc42Q61L mutant are 0.3 and 0.5 μM in GDP binding assay, respectively. Use as a molecular probe .
|
-

-
- HY-19617A
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-1 hydrochloride is an orally active and irreversible L858R/T790M mutant selective EGFR inhibitor. EGFR-IN-1 hydrochloride potently inhibits Gefitinib-resistant EGFR L858R, T790M with 100-fold selectivity over wild-type EGFR. EGFR-IN-1 hydrochloride displays strong antiproliferative activity against the H1975 cells and the first line mutant HCC827 cells. Antitumor activity .
|
-

-
- HY-13810
-
|
|
Raf
|
Cancer
|
|
PF-04880594 is a potent and selective RAF inhibitor. PF-04880594 inhibits both wild-type and mutant BRAF and CRAF. PF-04880594 shows antitumor activity .
|
-

-
- HY-168876
-
|
|
EGFR
|
Cancer
|
|
AZ14240475 is a potent, selective, and brain-penetrant inhibitor of EGFR Ex20Ins mutants, with the pIC50 of 7.6. AZ14240475 plays an important role in cancer research .
|
-

-
- HY-135805A
-
|
|
EGFR
|
Cancer
|
|
(Rac)-JBJ-04-125-02 is the racemate of JBJ-04-125-02. JBJ-04-125-02 is a potent, mutant-selective, allosteric and orally active EGFR inhibitor .
|
-

-
- HY-13237
-
-

-
- HY-19617B
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-1 TFA is an orally active and irreversible L858R/T790M mutant selective EGFR inhibitor. EGFR-IN-1 TFA potently inhibits Gefitinib-resistant EGFR L858R, T790M with 100-fold selectivity over wild-type EGFR. EGFR-IN-1 TFA displays strong antiproliferative activity against the H1975 cells and the first line mutant HCC827 cells. Antitumor activity .
|
-

-
- HY-19617
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-1 (compound 24) is an orally active and irreversible L858R/T790M mutant selective EGFR inhibitor. EGFR-IN-1 potently inhibits Gefitinib-resistant EGFR L858R, T790M with 100-fold selectivity over wild-type EGFR. EGFR-IN-1 displays strong antiproliferative activity against the H1975 cells and the first line mutant HCC827 cells. Antitumor activity .
|
-

-
- HY-112256
-
ACP-105
5 Publications Verification
|
Androgen Receptor
|
Inflammation/Immunology
|
|
ACP-105 is an orally available, selective amd potent androgen receptor modulator (SARM), with pEC50s of 9.0 and 9.3 for AR wild type and T877A mutant, respectively.
|
-

-
- HY-164315
-
|
|
Ras
|
Cancer
|
|
KRAS G12C inhibitor 67 (example 35) is a KRAS G12C inhibitor that selectively inhibits the growth of many KRAS G12C mutant tumor cell lines .
|
-

-
- HY-130253
-
|
|
IRAK
|
Cancer
|
|
IRAK4-IN-6 is an orally efficacious and selective IRAK4 inhibitor with an IC50 of 4 nM, and targetes MyD88 L265P mutant diffuse large B cell lymphoma .
|
-

- HY-163636
-
|
|
Phosphodiesterase (PDE)
Ras
|
Cancer
|
|
Deltaflexin3 is a potent PDE6D inhibitor. Deltaflexin3 reduces the signaling of Ras and selectively decreases the KRAS mutant and PDE6D-dependent cancer cells growth .
|
-

- HY-121708
-
|
|
c-Kit
|
Cancer
|
|
KI-328 is a novel inhibitor targeting KIT kinase that has selective activity against some KIT mutant kinases commonly found in acute myeloid leukemia (AML). KI-328 showed specificity for KIT kinase in in vitro kinase assays and inhibited the growth of wild-type (Wt) and mutant KIT-expressing cells, but had lower activity against D816V-KIT. Comparative analysis of the inhibitory effects of several potent KIT inhibitors on the growth of multiple mutant KIT-expressing cells showed that the multi-kinase inhibitors had comparable activity against D816V-KIT as against other mutant KITs; however, heat shock protein 90 (HSP90) inhibitors showed significant activity against D816V-KIT, inhibiting the growth of D816V-KIT-expressing cells at concentrations that did not affect the growth of other mutant KIT-expressing cells. These results suggest that potent KIT inhibitors have different activities against different types of KIT mutant kinases. Therefore, in clinical development, KIT inhibitors need to validate their activity against multiple types of KIT mutant kinases.
|
-

- HY-108887
-
|
|
Raf
|
Cancer
|
|
Anticancer agent 124 is an orally active, highly selective and potent pan RAF inhibitor. Anticancer agent 124 inhibits MAPK signalling in BRAF V600E, NRAS and KRAS mutant tumor cells .
|
-

- HY-18690
-
|
AG-221
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Enasidenib is an oral, potent, reversible, selective inhibitor of the IDH2 mutant enzymes, with IC50s of 100 and 400 nM against IDH2 R140Q and IDH2 R172K, respectively.
|
-

- HY-135914
-
|
|
EGFR
|
Cancer
|
|
JBJ-02-112-05 is a potent, mutant-selective, allosteric and orally active EGFR inhibitor with an IC50 of 15 nM for EGFR L858R/T790M .
|
-

- HY-124153
-
|
|
Btk
|
Cardiovascular Disease
Inflammation/Immunology
|
|
GNE-431 is a potent, selective and noncovalent “pan-BTK” inhibitor against C481R, T474I and T474Ms mutants. GNE-431 shows potency against wild-type BTK (IC50= 3.2 nM) and potency against C481S mutant (IC50= 2.5 nM). GNE-431 is proming for rasearch of haematological disorders and autoimmune diseases .
|
-

- HY-124761
-
|
|
Polo-like Kinase (PLK)
Autophagy
|
Cancer
|
|
Poloppin is a potent, cell penetrant inhibitor of the mitotic Polo-like kinase (PLK) (IC50=26.9 μM) and prevents the protein-protein interaction via the Polo-box domain (PBD) (Kd= 29.5 μM). Poloppin selectively kills cells expressing mutant KRAS, enhancing death in mitosis. Poloppin is used for the study of KRAS-mutant cancers as single agents, or in combination with c-MET inhibitors .
|
-

- HY-131312A
-
|
LY3410738 (gentisate); mutant IDH1-IN-6 (gentisate)
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Crelosidenib (gentisate) is a potent, selective and orally active mutant isocitrate dehydrogenase (IDH) inhibitor with IC50 of 6.27 nM, 3.71 nM, 36.9 nM and 11.5 nM for IDH1 R132H, IDH1 R132C, IDH2 R140Q and IDH2 R172K mutant enzymes, respectively. Crelosidenib (gentisate) is less active at inhibiting the IDH wild-type enzymes .
|
-

- HY-146243
-
|
|
Ras
Apoptosis
|
Cancer
|
|
TH-Z835 is a mutant selective KRAS (G12D) inhibitor with an IC50 of 1.6 μM. TH-Z835 inhibits both mantGMPPNP/GPPNP exchange and GPPNP/mantGMPPNP exchange .
|
-

- HY-145594
-
|
AB-291; DS-1001
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Safusidenib (AB-291; DS-1001) is an orally bioavailable, selective mutant IDH1 inhibitor. Safusidenib strongly inhibits mutant IDH1 but not wild-type IDH1. Safusidenib impairs tumor activity in chondrosarcoma . Safusidenib exhibits activity against IDH1R132H, and IDH1R132C with IC50s of 15, and 130 nM in assays without any preincubation, respectively .
|
-

- HY-13223A
-
|
CP 868596-26
|
Autophagy
PDGFR
FLT3
|
Cancer
|
|
Crenolanib benzenesulfonate is a potent and selective inhibitor of wild-type and mutant isoforms of the class III receptor tyrosine kinases FLT3 and PDGFRα/β with Kds of 0.74 nM and 2.1 nM/3.2 nM, respectively.
|
-

- HY-13223
-
|
CP-868596
|
FLT3
PDGFR
Autophagy
|
Cancer
|
|
Crenolanib is a potent and selective inhibitor of wild-type and mutant isoforms of the class III receptor tyrosine kinases FLT3 and PDGFRα/β with Kds of 0.74 nM and 2.1 nM/3.2 nM, respectively.
|
-

- HY-109061
-
|
YH25448; GNS-1480
|
EGFR
|
Cancer
|
|
Lazertinib (YH25448) is a potent, highly mutant-selective, blood-brain barrier permeable, orally available and irreversible third-generation EGFR tyrosine kinase inhibitor, and can be used in the research of non-small cell lung cancer .
|
-

- HY-139047
-
|
|
GLUT
|
Cancer
|
|
SW157765 is a selective non-canonical glucose transporter GLUT8 (SLC2A8) inhibitor. KRAS/KEAP1 double mutant NSCLC cells are selectively sensitive to the SW157765, due to the convergent consequences of dual KRAS and NRF2 modulation of metabolic and xenobiotic gene regulatory programs .
|
-

- HY-153306A
-
|
|
PI3K
Drug Isomer
|
Cancer
|
|
(S)-RLY-2608 is an S-enantiomer of of RLY-2608 (HY-153306). RLY-2608 is an orally active first-in-class allosteric mutant-selective inhibitor of PI3Ka with anti-tumor activity .
|
-

- HY-142161
-
ABD957
1 Publications Verification
|
MAGL
|
Cancer
|
|
ABD957 is a potent and selective covalent inhibitor of the ABHD17 family of depalmitoylases, with an IC50 of 0.21 µM for ABHD17B. ABD957 can block N-Ras signaling and the growth of NRAS-mutant AML cells .
|
-

- HY-156681
-
|
|
PI3K
|
Cancer
|
|
STX-478 (compound 80) is an oral CNS-penetrant allosteric mutant-selective PI3Kα inhibitor. STX-478 shows robust and durable tumor regression and can be used in cancer research .
|
-

- HY-129550
-
|
|
EGFR
|
Cancer
|
|
BI-4020 is a fourth-generation, orally active, and non-covalent EGFR tyrosine kinase inhibitor. BI-4020 inhibits not only the triple mutant EGFR del19 T790M C797S variant (IC50=0.2 nM in BaF3 cell lines) but also the double mutant EGFR del19 T790M and primary mutant EGFR del19 (IC50=1 nM). BI-4020 also shows activity against EGFR wt (IC50=190 nM). BI-4020 shows high kinome selectivity and good DMPK properties .
|
-

- HY-116572
-
|
|
APC
|
Cancer
|
|
TASIN-1 hydrochloride is a selective inhibitor of truncated APC gene (adenomatous polyposis coli gene) and exerts cytotoxic effects through inhibition of cholesterol biosynthesis. TASIN-1 hydrochloride represents a potential strategy for prevention and intervention in CRC with mutant APC .
|
-

- HY-15734
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
AGI-6780 that potently and selectively inhibits the tumor-associated mutant IDH2 R140Q with IC50 of 23±1.7 nM. AGI-6780 is less potent against IDH2 WT with IC50 of 190±8.1 nM.
|
-

- HY-164464
-
|
|
EGFR
|
Cancer
|
|
BPI-15086 an orally active, potent, irreversible mutant-selective inhibitor of both EGFR and T790M resistance mutations tyrosine kinase. BPI-15086 can be used for the research of non-small-cell lung cancer .
|
-

- HY-101522
-
|
|
EGFR
BMX Kinase
Btk
MEK
|
Cancer
|
|
CHMFL-EGFR-202 is a potent, irreversible inhibitor of epidermal growth factor receptor (EGFR) mutant kinase, with IC50s of 5.3 nM and 8.3 nM for drug-resistant mutant EGFR T790M and WT EGFR kinases, respectively. CHMFL-EGFR-202 exhibits ~10-fold selectivity for EGFR L858R/T790M against the EGFR wild-type in cells. CHMFL-EGFR-202 adopts a covalent “DFG-in-C-helix-out” inactive binding conformation with EGFR, with strong antiproliferative effects against EGFR mutant-driven nonsmall-cell lung cancer (NSCLC) cell lines .
|
-

- HY-145514
-
|
|
PROTACs
|
Cancer
|
|
dTAGV-1 TFA is a potent and selective degrader of mutant FKBP12 F36V fusion proteins. dTAGV-1 TFA can induce degradation of FKBP12 F36V-Nluc in vivo .
|
-

- HY-173155
-
|
|
Akt
|
Cancer
|
|
AKT1-IN-9 (4) is a selective mutant AKT1(E17K) inhibitor, with EC50 values of 9 nM and 995 nM in LAPC4-CR and SkBr3 cells, respectively .
|
-

- HY-116572A
-
|
|
APC
|
Cancer
|
|
TASIN-1 is a selective inhibitor of truncated APC gene (adenomatous polyposis coli gene) and exerts cytotoxic effects through inhibition of cholesterol biosynthesis. TASIN-1 represents a potential therapeutic strategy for prevention and intervention in CRC with mutant APC .
|
-

- HY-139997
-
|
|
PROTACs
EGFR
|
Cancer
|
|
DDC-01-163 is a mutant-selective allosteric PROTAC-based EGFR degrader, with an IC50 of 45 nM (Pink: EGFR ligand (HY-151048); Black: linker (HY-W008005); Blue: CRBN ligand (HY-14658)) .
|
-

- HY-114778
-
|
SHR3162; Fuzuloparib
|
PARP
|
Cancer
|
|
Fluzoparib (SHR3162) is a potent and orally active PARP1 inhibitor (IC50=1.46±0.72 nM, a cell-free enzymatic assay) with superior antitumor activity. Fluzoparib selectively inhibits the proliferation of homologous recombination repair (HR)-deficient cells, and sensitizes both HR-deficient and HR-proficient cells to cytotoxic agents. Fluzoparib exhibits good pharmacokinetic properties in vivo and can be used for BRCA1/2-mutant relapsed ovarian cancer research .
|
-

- HY-10358
-
|
MK-2206 (2HCl)
|
Organoid
Akt
Autophagy
Apoptosis
|
Cancer
|
|
MK-2206 dihydrochloride (MK-2206 (2HCl)) is an orally active, BBB-penetrated allosteric AKT inhibitor with IC50s of 5 nM, 12 nM, and 65 nM for AKT1, AKT2, and AKT3, respectively. MK-2206 dihydrochloride induces autophagy .
|
-

- HY-79077
-
|
AZD-9291 dimesylate; Mereletinib dimesylate
|
EGFR
|
Cancer
|
|
Osimertinib dimesylate (AZD-9291 dimesylate) is an irreversible and mutant selective EGFR inhibitor with IC50s of 12 and 1 nM against EGFR L858R and EGFR L858R/T790M, respectively.
|
-

- HY-164488
-
|
|
PAK
|
Cancer
|
|
KT D606 is an inhibitor of the PAK kinase family, with an IC50 value of 4 μM. KT D606 selectively blocks the proliferation of cancer cells transformed by oncogenic RAS mutants and can be used for research on RAS/PAK1-induced cancers .
|
-

- HY-110392
-
|
|
Estrogen Receptor/ERR
|
Cancer
|
|
CMP8, a selective ligand for estrogen receptor, binds to the mutant estrogen receptor ligand binding domain (ERLBD). CMP8 exhibits IC50 values of 29 nM , 41 nM, 1100 nM and 2200 nM for MGERα, MGRERα, hERα and hERβ, respectively .
|
-

- HY-100214
-
|
|
EGFR
|
Cancer
|
|
EAI001 is a potent, selective mutant epidermal growth factor receptor (EGFR) allosteric inhibitor with an IC50 value of 24 nM for EGFR L858R/T790M. EAI001 can be used for research of cancer .
|
-

- HY-103682A
-
|
|
Histone Methyltransferase
|
Cancer
|
|
PF-06726304 acetate is a potent and selective EZH2 inhibitor. PF-06726304 acetate inhibits wild-type and Y641N mutant EZH2 with Kis of 0.7 and 3.0 nM, respectively. PF-06726304 acetate displays robust antitumor growth activity .
|
-

- HY-103682
-
|
|
Histone Methyltransferase
|
Cancer
|
|
PF-06726304 is a potent and selective EZH2 inhibitor. PF-06726304 inhibits wild-type and Y641N mutant EZH2 with Kis of 0.7 and 3.0 nM, respectively. PF-06726304 displays robust antitumor growth activity .
|
-

- HY-112306
-
|
DCC-2618
|
c-Kit
PDGFR
FLT3
VEGFR
Apoptosis
|
Cancer
|
|
Ripretinib (DCC-2618) is an orally bioavailable, selective KIT and PDGFRA switch-control inhibitor. Ripretinib (DCC-2618) targets and binds to both wild-type and mutant forms of KIT and PDGFRA specifically at their switch pocket binding sites, thereby preventing the switch from inactive to active conformations of these kinases and inactivating their wild-type and mutant forms. Ripretinib (DCC-2618) also inhibits multiple other kinase targets, such as FLT3 and KDR (or VEGFR-2) . DCC-2618 exerts antineoplastic effect and induces apoptosis .
|
-

- HY-154959A
-
|
|
Ras
|
Cancer
|
|
(9R,12aR)-AZD4747 is a diastereomer of AZD4747 (HY-154959). AZD4747 is a selective mutant GTPase KRAS G12C inhibitor with blood-brain barrier permeability. AZD4747 has the potential to study cancer .
|
-

- HY-12343
-
|
CID-53347902
|
Potassium Channel
|
Cardiovascular Disease
|
|
ML277 (CID-53347902) is a potent and selective activator of K(v)7.1 (KCNQ1) potassium channel activator (EC50=270 nM), rescues function of pathophysiologically important mutant channel complexes in human induced pluripotent stem cell-derived cardiomyocytes .
|
-

- HY-144605
-
|
|
EGFR
PROTACs
|
Cancer
|
|
PROTAC EGFR degrader 3 is a potent PROTAC EGFR degrader. PROTAC EGFR degrader 3 shows excellent cellular activity against the H1975 and HCC827 cells with high selectively. PROTAC EGFR degrader 3 shows that the lysosome is involved in the degradation process of EGFR mutant degradation .
|
-

- HY-101119
-
|
|
Estrogen Receptor/ERR
|
Cancer
|
|
GLL398 is a potent, orally bioavailable selective estrogen receptor degrader (SERD) with an IC50 value of 1.14 nM. GLL398 shows a strong dose-dependent binding to ER with a mutation at Y537S (IC50=29.5 nM). GLL398 blocks tumor growth in xenograft models of breast cancer.
|
-

- HY-151523
-
|
|
Ras
|
Cancer
|
|
KRas G12R inhibitor 1 (compound 3) is a KRas G12R selective covalent inhibitor that exploits the strong nucleophilicity of mutant cysteines and binds irreversibly in the Switch II region of K-Ras. KRas G12R inhibitor 1 can be used in cancer research .
|
-

- HY-123824
-
|
|
Sodium Channel
|
Neurological Disease
|
|
GNE-0439 is a novel Nav1.7-selective inhibitor with IC50 of 0.34 uM and inhibits Nav1.5 with an IC50 of 38.3 μM. GNE-0439 inhibits mutant N1742K channels (IC50=0.37 uM) in membrane potential assays. GNE-0439 possesses a carboxylic acid group, binds outside of the channel pore, and is unique compared with known selective VSD4 binders .
|
-

- HY-143319
-
|
|
EGFR
|
Cancer
|
|
SPH5030 is a selective and irreversible HER2 inhibitor. SPH5030 inhibits HER2 WT and EGFR WT with IC50s of 3.51 and 8.13 nM, respectively. SPH5030 shows excellent activities against HER2 mutants. SPH5030 can be used for the research of cancer .
|
-

- HY-157287
-
|
|
Others
|
Inflammation/Immunology
|
|
(S)-Phe-A110/B319 is a selective binde that has 20-fold higher affinity towards the H1047R mutant of p110α in the p110α/p85α PI3K complex .
|
-

- HY-110094
-
|
|
5-HT Receptor
|
Neurological Disease
|
|
BIMU 8 is a potent and selective 5-HT4 agonist with EC50s of 18 nM, 77 nM, and 540 nM for wild type 5HT4 receptor, T3.36A, and W6.48A mutant 5-HT4 receptors .
|
-

- HY-114043
-
|
|
Glycosidase
|
Metabolic Disease
|
|
NCGC00092410 is a potent, selective, and nonsugar glucocerebrosidase (GC) inhibitor, with an IC50 of 31 nM. NCGC00092410 shows no activity against the related hydrolases at concentrations up to 77 μM. NCGC00092410, a GC chaperone, and increases the activity and lysosomal localization of glucocerebrosidase in mutant cell lines. NCGC00092410 can be used for the research of Gaucher disease .
|
-

- HY-W740378
-
|
AFN-941
|
EGFR
JAK
|
Others
|
|
1,2,3,4-Tetrahydrostaurosporine is a derivative of Staurosporine (HY-15141) and an inhibitor of mutant EGFR (IC50=74 nM for EGFRT790M). It is selective for EGFRT790M over wild-type EGFR (IC50=390 nM). It also binds to Janus kinase 3 (JAK3).
|
-

- HY-N12390
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Alternaphenol B2 is a selective IDH1 inhibitor from the coral-derived fungus Parengyodontium album SCSIO SX7W11. Alternaphenol B2 shows inhibitory activity against isocitrate dehydrogenase mutant R132H (IDH1m), with IC50 values of 41.9 μM .
|
-

- HY-101494
-
|
LY3214996
|
ERK
|
Cancer
|
|
Temuterkib (LY3214996) is a highly selective inhibitor of ERK1 and ERK2, with IC50 of 5 nM for both enzymes in biochemical assays. Temuterkib potently inhibits cellular p-RSK1 in BRAF and RAS mutant cancer cell lines. Temuterkib shows potent antitumor activities in cancer models with MAPK pathway alterations.
|
-

- HY-139590
-
|
BOS-172738; DS-5010
|
RET
PDGFR
|
Cancer
|
|
Zeteletinib (BOS-172738; DS-5010) is an orally active, selective RET kinase inhibitor with nanomolar potency against RET and >300-fold selectivity against VEGFR2. Zeteletinib shows exquisite potency for the wild type RET, RET V804M/L gatekeeper mutants, and the most common oncogenic RET mutation M918T. Zeteletinib has potent antitumor activity .
|
-

- HY-134813
-
|
|
Ras
|
Cancer
|
|
MRTX1133 is a noncovalent, potent, and selective alkyne-based KRAS G12D inhibitor. MRTX1133 optimally fills the switch II pocket and extends three substituents to favorably interact with the protein, resulting in an estimated KD against KRAS G12D of 0.2 pM. MRTX1133 prevents SOS1-catalyzed nucleotide exchange and/or formation of the KRAS G12D/GTP/RAF1 complex, thereby inhibiting mutant KRAS-dependent signal transduction. MRTX1133 selectively inhibits KRAS G12D mutant, but not KRAS wild-type, tumor cells. MRTX1133 has single digit nanomolar activity in cellular assays and marked in vivo efficacy in tumor models harboring KRAS G12D mutations .
|
-

- HY-143234
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
mIDH1-IN-1 (compound 43) is a potent and selective mIDH1 (mutant isocitrate dehydrogenases 1) inhibitor, with an IC50 of 961.5 nM. mIDH1-IN-1 potently inhibits intracellular 2-HG (2-hydroxyglutarate) production in HT1080 cells, with an EC50 of 208.6 ± 8.0 nM. mIDH1-IN-1 shows a significant anti-proliferation activity on IDH1 mutant-U-87 cells, with an IC50 of 41.8 nM. mIDH1-IN-1 is an antitumor agent, and can be used for IDH1 mutated solid tumors research .
|
-

- HY-141523
-
|
RMC-4630; SHP2-IN-7
|
SHP2
Phosphatase
|
Cancer
|
|
Vociprotafib (RMC-4630) is an orally active, selective and potent phosphatase SHP2 inhibitor, which blocks activation of the RAS-RAF-MEK-ERK signaling pathway with antitumor activity. Vociprotafib accelerates the time to, and increases the magnitude of, tumor regressions in Osimertinib (HY-15772)-sensitive EGFR-mutant tumors of mice .
|
-

- HY-136938
-
NEO2734
1 Publications Verification
EP31670
|
Epigenetic Reader Domain
Histone Acetyltransferase
|
Cancer
|
|
NEO2734 (EP31670) is an orally active dual p300/CBP and BET bromodomain selective inhibitor, with IC50 values of <30 nM for both p300/CBP and BET bromodomains . NEO2734 is active in SPOP mutant and wild-type prostate cancer .
|
-

- HY-155285
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
YS-363 is a potent, selective, and orally active EGFR inhibitor, with IC50s of 0.96 nM and 0.67 nM for wild-type and L858R mutant forms of EGFR, respectively. YS-363 can induce G0/G1 cell cycle arrest and apoptosis .
|
-

- HY-145702
-
|
|
MEK
ERK
|
Cancer
|
|
MAP855 is a highly potent, selective, ATP-competitive and orally active MEK1/2 kinase inhibitor (MEK1 ERK2 cascade IC50=3 nM, pERK EC50=5 nM). MAP855 shows equipotent inhibition of wild-type and mutant MEK1/2 .
|
-

- HY-128784
-
|
|
MDM-2/p53
Reactive Oxygen Species
|
Cancer
|
|
PK11007 is a mild thiol alkylator with anticancer activity. PK11007 stabilizes p53 via selective alkylation of two surface-exposed cysteines without compromising its DNA binding activity. PK11007 induces mutant p53 cancer cell death by increasing reactive oxygen species (ROS) levels .
|
-

- HY-15772S1
-
|
AZD-9291-13C,d3; Mereletinib-13C,d3
|
EGFR
|
Cancer
|
|
Osimertinib- 13C,d3 is the deuterium and 13C labeled Osimertinib. Osimertinib (AZD9291) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M, respectively.
|
-

- HY-139590A
-
|
BOS-172738 hemiadipate; DS-5010 hemiadipate
|
RET
PDGFR
|
Cancer
|
|
Zeteletinib (BOS-172738; DS-5010) hemiadipate is an orally active, selective RET kinase inhibitor with nanomolar potency against RET and >300-fold selectivity against VEGFR2. Zeteletinib hemiadipate shows exquisite potency for the wild type RET, RET V804M/L gatekeeper mutants, and the most common oncogenic RET mutation M918T. Zeteletinib hemiadipate has potent antitumor activity .
|
-

- HY-101562
-
|
GDC-0077; RG6114
|
PI3K
Apoptosis
|
Cancer
|
|
Inavolisib (GDC-0077) is a potent, orally active, and selective PI3Kα inhibitor (IC50=0.038 nM). Inavolisib exerts its activity by binding to the ATP binding site of PI3K, thereby inhibiting the phosphorylation of PIP2 to PIP3. Inavolisib is more selective for mutant versus wild-type PI3Kα. Inavolisib can be used for the study of breast cancer .
|
-

- HY-114436
-
|
|
Ras
|
Cancer
|
|
MRTX-1257 is a selective, irreversible, covalent and orally active KRAS G12C inhibitor, with an IC50 of 900 pM for KRAS dependent ERK phosphorylation in H358 cells .
|
-

- HY-114436S
-
|
|
Isotope-Labeled Compounds
|
Cancer
|
|
MRTX-1257-d6 is the deuterium labeled MRTX-1257 (HY-114436). MRTX-1257 is a selective, irreversible, covalent and orally active KRAS G12C inhibitor, with an IC50 of 900 pM for KRAS dependent ERK phosphorylation in H358 cells .
|
-

- HY-136789
-
|
BDTX-189
|
EGFR
|
Cancer
|
|
Tuxobertinib (BDTX-189) is a potent, orally active and selective inhibitor of allosteric EGFR and HER2 oncogenic mutations, including EGFR/HER2 exon 20 insertion mutants. Tuxobertinib shows KDs of 0.2, 0.76, 13 and 1.2 nM for EGFR, HER2, BLK and RIPK2, reapectively. Anticancer activity .
|
-

- HY-112870
-
|
Alflutinib; Furmonertinib; AST2818
|
EGFR
|
Cancer
|
|
Firmonertinib (Alflutinib; Furmonertinib) is an orally active, mutant-selective, and highly brain penetrant EGFR inhibitor. Firmonertinib inhibits EGFR active mutations as well as the T790M acquired resistant mutation. Firmonertinib has the potential for the research of cancer diseases, especially advanced non-small cell lung cancer (NSCLC) with EGFR ex20ins mutation .
|
-

- HY-112601
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
IDH1 Inhibitor 1 is a potent, orally bioavailable, brain-penetrant and selective mutant IDH1 inhibitor with IC50s of 0.021 μM, 0.045 μM, and 2.52 μM for IDH1 R132H, IDH1 R132C, and IDH1 WT, respectively . Anticancer activity .
|
-

- HY-15103
-
|
|
PTHR
Androgen Receptor
|
Cancer
|
|
AH3960 (compound 16c) is an antagonist of androgen receptor. AH3960 binds wild as well as T877 mutant type androgen receptors. AH3960 selectively inhibits T877 with an IC50 value of 0.82 μM. AH3960 also serves as an agonist of parathyroid hormone receptor-1 (PTHR1) .
|
-

- HY-15772
-
|
AZD-9291; Mereletinib
|
EGFR
|
Cancer
|
|
Osimertinib (AZD9291) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M, respectively. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
|
-

- HY-112870A
-
|
Alflutinib mesylate; Furmonertinib mesylate; AST2818 mesylate
|
EGFR
|
Cancer
|
|
Firmonertinib (Alflutinib; Furmonertinib) mesylate is is an orally active, mutant-selective, and highly brain penetrant EGFR inhibitor. Firmonertinib mesylate inhibits EGFR active mutations as well as the T790M acquired resistant mutation. Firmonertinib mesylate has the potential for the research of cancer diseases, especially advanced non-small cell lung cancer (NSCLC) with EGFR ex20ins mutation .
|
-

- HY-15772A
-
|
AZD-9291 mesylate; Mereletinib mesylate
|
EGFR
|
Cancer
|
|
Osimertinib mesylate (AZD9291 mesylate) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
|
-

- HY-104066A
-
|
Xiliertinib tartrate; HMPL-309 tartrate
|
EGFR
|
Cancer
|
|
Theliatinib (Xiliertinib) tartrate is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib (tartrate) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-104066
-
|
Xiliertinib; HMPL-309
|
EGFR
|
Cancer
|
|
Theliatinib (Xiliertinib) is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-112301
-
|
BLU-667
|
RET
|
Cancer
|
|
Pralsetinib (BLU-667) is a highly potent, selective RET inhibitor. Pralsetinib (BLU-667) inhibits WT RET, RET mutants V804L, V804M, M918T and CCDC6-RET fusion with IC50s of 0.4, 0.3, 0.4, 0.4, and 0.4 nM, respectively .
|
-

- HY-121845
-
|
|
HSP
|
Metabolic Disease
|
|
4-Br-Bnlm is a selective inhibitor of glucose-regulated protein 94 (Grp94) with an EC50 value of 0.96 µM. 4-Br-Bnlm reduces the levels of mutant myocilin proteins as well as wild-type myocilin misfold in cells. 4-Br-Bnlm promotes the clearance of toxic formsof myocilin and reduces myocilin toxicity .
|
-

- HY-18690S
-
|
AG-221-d6
|
Isotope-Labeled Compounds
Isocitrate Dehydrogenase (IDH)
|
Others
|
|
Enasidenib-d6 (AG-221-d6) is the deuterium labeled Enasidenib (HY-18690) . Enasidenib is an oral, potent, reversible, selective inhibitor of the IDH2 mutant enzymes, with IC50s of 100 and 400 nM against IDH2 R140Q and IDH2 R172K, respectively .
|
-

- HY-146223A
-
|
|
Ras
|
Cancer
|
|
(3R,10R,14aS)-AZD4625 is the isomer of AZD4625 (HY-146223), and can be used as an experimental control. AZD4625 (Compound 21) is a highly potent, selective, covalent and allosteric inhibitor of the mutant GTPase KRAS G12C. AZD4625 has high oral bioavailability .
|
-

- HY-147941
-
|
|
PROTACs
EGFR
|
Cancer
|
|
MS9427 is a potent PROTAC EGFR degrader with Kds of 7.1 nM and 4.3 nM for EGFR WT and EGFR L858R, respectively. MS9427 selectively degrades the mutant but not the WT EGFR through both the ubiquitin/proteasome system (UPS) and autophagy/lysosome pathways. MS9427 potently inhibits the proliferation of NSCLC cells. MS9427 can be used for researching anticancer .
|
-

- HY-130608
-
|
|
EGFR
|
Cancer
|
|
Mutated EGFR-IN-3 (compound 3) is a potent, ATP-competitive and highly selective allosteric dibenzodiazepinone inhibitor of the EGFR(L858R/T790M) and EGFR(L858R/T790M/C797S) mutants with IC50 values of 12 nM and 13 nM, respectively .
|
-

- HY-14174
-
|
|
γ-secretase
|
Neurological Disease
Cancer
|
|
MRK-560 is an orally active, brain barrier-penetrated γ-Secretase inhibitor, reducing Aβ peptide in rat brain and cerebrospinal fluid. MRK-560 decreases mutant NOTCH1 processing by selectively inhibiting PSEN1. MRK-560 can be used in studies of Alzheimer's disease and T-cell acute lymphoblastic leukaemia (T-ALL) .
|
-

- HY-12026
-
|
|
EGFR
|
Cancer
|
|
WZ4002 is a mutant selective EGFR inhibitor with IC50s of 2, 8, 3 and 2 nM for EGFR L858R, EGFR L858R/T790M, EGFR E746_A750 and EGFR E746_A750/T790M, respectively.
|
-

- HY-153663
-
|
|
Ras
|
Cancer
|
|
TH-Z827 is a mutant selective KRAS(G12D) inhibitor with an IC50 of 2.4 μM. TH-Z827 does not bind KRAS(WT) or KRAS(G12C). TH-Z827 blocked the KRAS(G12D)-CRAF interaction with an IC50 value of 42 μM .
|
-

- HY-12755
-
|
CID-2950007
|
Ras
Apoptosis
|
Cancer
|
|
ML141 (CID-2950007) is a potent, allosteric, selective and reversible non-competitive inhibitor of Cdc42 GTPase. ML141 inhibits Cdc42 wild type and Cdc42 Q61L mutant with EC50s of 2.1 and 2.6 μM, respectively. ML141 shows low micromolar potency and selectivity against other members of the Rho family of GTPases (Rac1, Rab2, Rab7). ML141 do not show cytotoxicity in multiple cell lines .
|
-

- HY-17499
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
EGFR-IN-12 is a 4,6-disubstituted pyrimidine and is a potent, ATP-competitive, irreversible and highly selective EGFR inhibitor with an IC50of 21 nM. EGFR-IN-12 also inhibits mutant EGFR L858R and EGFR L861Q with IC50s of 63 nM and 4 nM, respectively. EGFR-IN-12 displays strong selectivity for EGFR over HER4 (IC50 = 7640 nM) and a panel of 55 other kinases. EGFR-IN-12 induces cells apoptosis and has antitumor activity .
|
-

- HY-12001
-
|
|
EGFR
|
Cancer
|
|
WZ3146 is a mutant selective EGFR inhibitor with IC50s of
2, 2, 5, 14 and 66 nM for EGFR L858R, EGFR L858R/T790M, EGFR E746_A750, EGFR E746_A750/T790M and EGFR, respectively.
|
-

- HY-116804
-
|
|
Histone Methyltransferase
|
Cancer
|
|
ZLD1039 is a potent, highly selective, and orally bioavailable EZH2 inhibitor. ZLD1039 shows potent and concentration-dependent inhibition of PRC2 enzymatic activity against EZH2 wild-type as well as Y641F, and A677G mutant enzymes with IC50 values of 5.6, 15, and 4.0 nM, respectively. ZLD1039 inhibits breast tumor growth and metastasis .
|
-

- HY-108232
-
MK-2206
Maximum Cited Publications
368 Publications Verification
|
Organoid
Akt
Autophagy
Apoptosis
|
Cancer
|
|
MK-2206 is an orally active, highly potent and selective allosteric Akt inhibitor, with IC50s of 8, 12, and 65 nM for Akt1, Akt2, and Akt3, respectively. Many breast cancer cell lines, and PIK3CA-mutant and cell lines with PTEN loss are sensitive to MK-2206. MK-2206 has anticancer activities .
|
-

- HY-128862
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-7 is a potent, selective and orally active EGFR kinase inhibitor. EGFR-IN-7 has inhibitory effect for for EGFR (WT) and EGFR (mutant C797S/T790M/L858R) with IC50 values of 7.92 nM and 0.218 nM, respectively. EGFR-IN-7 can be used for the research of various cancers .
|
-

- HY-19834
-
|
GDC-0853
|
Btk
|
Cancer
|
|
Fenebrutinib (GDC-0853) is a potent, selective, orally available, and noncovalent bruton's tyrosine kinase (Btk) inhibitor with Kis of 0.91 nM, 1.6, 1.3, 12.6, and 3.4 nM for WT Btk, and the C481S, C481R, T474I, T474M mutants. Fenebrutinib has the potential for rheumatoid arthritis and systemic lupus erythematosus research .
|
-

- HY-15772S
-
|
AZD-9291-d6; Mereletinib-d6
|
EGFR
|
Cancer
|
|
Osimertinib-d6 is a deuterium labeled osimertinib. Osimertinib is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer[1].
|
-

- HY-114226
-
|
FT-2102
|
Isocitrate Dehydrogenase (IDH)
|
Inflammation/Immunology
Cancer
|
|
Olutasidenib (FT-2102) is a highly potent, orally active, brain penetrant and selective inhibitor of mutant Isocitrate dehydrogenase 1 (IDH1), with IC50 values of 21.2 nM and 114 nM for IDH1- R132H and IDH1- R132C, respectively . Olutasidenib (FT-2102) is under the study in the treatment of acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS) .
|
-

- HY-142160
-
|
|
Raf
|
Cancer
|
|
GNE-9815 (compound 7) is a highly selective, pan-RAF inhibitor with good oral bioavailability. GNE-9815 exhibits Ki values of 0.062 and 0.19 nM for CRAF and BRAF, respectively. GNE-9815 combines with MEK inhibitor Cobimetinib (HY-13064) shows synergistic modulation of MAPK pathway. GNE-9815 can be used in studies of KRAS mutant cancers .
|
-

- HY-19985A
-
|
|
EGFR
|
Cancer
|
|
(3S, 4S)-PF-06459988 is the S enantiomer of PF-06459988 with less active. PF-06459988 is a potent irreversible inhibitor of T790M mutant epidermal growth factor receptor (EGFR). PF-06459988 has excellent selectivity against EGFR wild-type while possessing a minimally reactive electrophile that reduces the propensity of off-target labeling .
|
-

- HY-114358
-
|
ONO-7475
|
TAM Receptor
Trk Receptor
|
Cancer
|
|
Tamnorzatinib (ONO-7475) is a potent, selective, and orally active Axl/Mer inhibitor with IC50 values of 0.7 nM and 1.0 nM, respectively. Tamnorzatinib sensitizes AXL-overexpressing EGFR-mutant NSCLC cells to the EGFR-TKIs, suppresses the emergence and maintenance of tolerant cells. Tamnorzatinib combines with Osimertinib (HY-15772) provides a bright promise for the study of EGFR-mutated non-small cell lung cancer (NSCLC).
|
-

- HY-14588
-
|
ABT-378
|
HIV
HIV Protease
SARS-CoV
|
Infection
Cancer
|
|
Lopinavir (ABT-378) is a highly potent, selective peptidomimetic inhibitor of the HIV-1 protease, with Kis of 1.3 to 3.6 pM for wild-type and mutant HIV protease. Lopinavir acts by arresting maturation of HIV-1 thereby blocking its infectivity . Lopinavir is also a SARS-CoV 3CL pro inhibitor with an IC50 of 14.2 μM .
|
-

- HY-151903S
-
|
|
FGFR
|
Cancer
|
|
FGFR2/3-IN-1 is a potent and selective FGFR2 and FGFR3 (FGFR) inhibitor with IC50s of 1 nM and 0.5 nM, respectively. FGFR2/3-IN-1 displays >40-fold selectivity over FGFR1/FGFR4 and other kinome. FGFR2/3-IN-1 also inhibits FGFR3 V555L and V555M mutants with IC50s of 2.7 nM and 6.1 nM, respectively[1].
|
-

- HY-10683
-
|
|
PI3K
mTOR
|
Cancer
|
|
PKI-402 is a selective, reversible, ATP-competitive inhibitor of PI3K, including PI3K-α mutants, and mTOR (IC50=2, 3, 7,14 and 16 nM for PI3Kα, mTOR, PI3Kβ, PI3Kδ and PI3Kγ).
|
-

- HY-145574
-
|
WX-0593
|
Anaplastic lymphoma kinase (ALK)
ROS Kinase
BCRP
Akt
ERK
STAT
|
Cancer
|
|
Iruplinalkib (WX-0593) is an orally active and selective ALK/ROS1 inhibitor. Iruplinalkib can effectively inhibit tyrosine autophosphorylation of ALK and mutant ALK, EGFR, with the IC50 between 5.38 and 16.74 nM. Iruplinalkib is also a suppressive agent of the transporter MATE1, MATE2K, P-gp and BCRP. Iruplinalkib can be used in the study of non-small cell lung cancer .
|
-

- HY-130259
-
|
|
Atg8/LC3
ATTECs
Autophagy
|
Neurological Disease
|
|
LC3-mHTT-IN-AN2 (Compound AN2) is a mHTT-LC3 linker compound, which interacts with both mutant huntingtin protein (mHTT) and LC3B but not with wtHTT or irrelevant control proteins. LC3-mHTT-IN-AN2 reduces the levels of mHTT in an allele-selective manner in cultured Huntington disease (HD) mouse neurons .
|
-

- HY-147941A
-
|
|
PROTACs
EGFR
|
Cancer
|
|
MS9427 TFA is a potent PROTAC EGFR degrader with Kds of 7.1 nM and 4.3 nM for EGFR WT and EGFR L858R, respectively. MS9427 TFA selectively degrades the mutant but not the WT EGFR through both the ubiquitin/proteasome system (UPS) and autophagy/lysosome pathways. MS9427 TFA potently inhibits the proliferation of NSCLC cells. MS9427 TFA can be used for researching anticancer .
|
-

- HY-146112
-
|
|
IRAK
|
Cancer
|
|
IRAK4-IN-14 (compound 28) is a potent, selective and orally active IRAK4 inhibitor with an IC50 of 0.003 µM. IRAK4-IN-14 shows good PK parameters in rats and mouse. IRAK4-IN-14 shows synergistic in vitro activity against MyD88/CD79 double mutant ABC-DLBCL in combination with Acalabrutinib .
|
-

- HY-130258
-
|
|
Atg8/LC3
ATTECs
Autophagy
|
Neurological Disease
|
|
LC3-mHTT-IN-AN1 (Compound AN1) is a mHTT-LC3 linker compound, which interacts with both mutant huntingtin protein (mHTT) and LC3B but not with wtHTT or irrelevant control proteins. LC3-mHTT-IN-AN1 reduces the levels of mHTT in an allele-selective manner in cultured Huntington disease (HD) mouse neurons .
|
-

- HY-130149
-
|
MRTX849
|
Ras
|
Cancer
|
|
Adagrasib (MRTX849) is a potent, orally-available, and mutation-selective covalent inhibitor of KRAS G12C with potential antineoplastic activity. Adagrasib covalently binds to KRAS G12C at the cysteine at residue 12, locks the protein in its inactive GDP-bound conformation, and inhibits KRAS-dependent signal transduction .
|
-
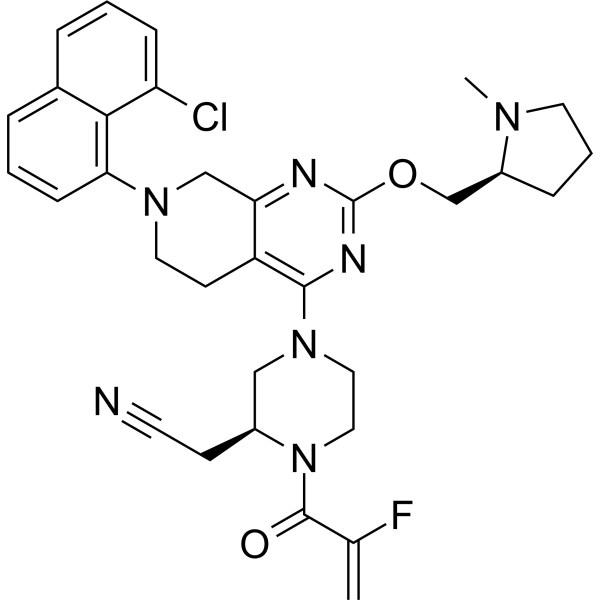
- HY-147183
-
|
|
EGFR
|
Cancer
|
|
JBJ-09-063 is a mutant-selective allosteric EGFR inhibitor with IC50s of 0.147 nM, 0.063 nM, 0.083 nM and 0.396 nM for EGFR L858R, EGFR L858R/T790M, EGFR L858R/T790M/C797S and EGFRLT/L747S. JBJ-09-063 effectively reduces EGFR, Akt and ERK1/2 phosphorylation. JBJ-09-063 is effective across EGFR tyrosine kinase inhibitor (TKI)-sensitive and resistant models. JBJ-09-063 can be used for researching EGFR-mutant lung cancer .
|
-

- HY-147183A
-
|
|
EGFR
|
Cancer
|
|
JBJ-09-063 TFA is a mutant-selective allosteric EGFR inhibitor with IC50s of 0.147 nM, 0.063 nM, 0.083 nM and 0.396 nM for EGFR L858R, EGFR L858R/T790M, EGFR L858R/T790M/C797S and EGFRLT/L747S. JBJ-09-063 TFA effectively reduces EGFR, Akt and ERK1/2 phosphorylation. JBJ-09-063 TFA is effective across EGFR tyrosine kinase inhibitor (TKI)-sensitive and resistant models. JBJ-09-063 TFA can be used for researching EGFR-mutant lung cancer .
|
-

- HY-147183B
-
|
|
EGFR
|
Cancer
|
|
JBJ-09-063 hydrochloride is a mutant-selective allosteric EGFR inhibitor with IC50s of 0.147 nM, 0.063 nM, 0.083 nM and 0.396 nM for EGFR L858R, EGFR L858R/T790M, EGFR L858R/T790M/C797S and EGFRLT/L747S. JBJ-09-063 hydrochloride effectively reduces EGFR, Akt and ERK1/2 phosphorylation. JBJ-09-063 hydrochloride is effective across EGFR tyrosine kinase inhibitor (TKI)-sensitive and resistant models. JBJ-09-063 hydrochloride can be used for researching EGFR-mutant lung cancer .
|
-

- HY-15772AR
-
|
|
EGFR
|
Cancer
|
|
Osimertinib (mesylate) (Standard) is the analytical standard of Osimertinib (mesylate). This product is intended for research and analytical applications. Osimertinib mesylate (AZD9291 mesylate) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
|
-

- HY-15772R
-
|
|
EGFR
|
Cancer
|
|
Osimertinib (Standard) is the analytical standard of Osimertinib. This product is intended for research and analytical applications. Osimertinib (AZD9291) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M, respectively. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
|
-

- HY-164487
-
|
|
Proteasome
|
Cancer
|
|
JBJ-08-178-01 is a mutant-selective tyrosine kinase inhibitor against human epidermal growth factor receptor 2 (HER2) with an antitumoral activity. JBJ-08-178-01 reduces both the kinase activity and protein levels of HER2 by inducing proteasomal degradation of the receptor in lung cancer. JBJ-08-178-01 is promising for research of non-small-cell lung cancer .
|
-

- HY-12972
-
|
PF-06747775
|
EGFR
|
Cancer
|
|
Mavelertinib is a selective, orally available and irreversible EGFR tyrosine kinase inhibitor (EGFR TKI), with IC50s of 5, 4, 12 and 3 nM for Del, L858R, and double mutants T790M/L858R and T790M/Del, respectively. Mavelertinib can be used for the research of non-small-cell lung cancer (NSCLC) .
|
-

- HY-146113
-
|
|
IRAK
|
Cancer
|
|
IRAK4-IN-15 (compound 35) is a potent and selective IRAK4 inhibitor with an IC50 of 0.002 µM. IRAK4-IN-15 shows good human PK predictions with low intrinsic clearance. IRAK4-IN-15 shows great synergistic in vitro activity against MyD88/CD79 double mutant ABC-DLBCL in combination with Acalabrutinib. .
|
-

- HY-158325
-
|
|
PROTACs
FLT3
Apoptosis
|
Cancer
|
|
PROTAC FLT-3 degrader 4 is an orally active CRBN-based FLT3-PROTAC degrader that potently induces FLT3-ITD degradation through the ubiquitin-proteasome system. PROTAC FLT-3 degrader 4 shows highly selective to FLT3-ITD mutant acute myeloid leukemia (AML) cells. (Blue: CRBN ligand, Black: linker; Pink: FLT3 inhibitor) .
|
-

- HY-149555
-
|
|
Eukaryotic Initiation Factor (eIF)
|
Neurological Disease
|
|
DNL343 is a potent, selective, orally active and brain-penetrant activator of eukaryotic initiation factor eIF2B. DNL343 inhibits the activity of the integrated stress response (ISR) in the central nervous system (CNS) and reverses neurodegeneration and neuroinflammation. DNL343 also prevents motor dysfunction and premature death in eIF2B loss-of-function (LOF) mutant mice. DNL343 can be used in the study of neurodegenerative diseases .
|
-

- HY-107779
-
|
|
Raf
|
Cancer
|
|
BI-882370 is a potent and selective RAF kinase inhibitor that binds to the ATP binding site of the kinase positioned in the DFG-out (inactive) conformation of the BRAF kinase. BI-882370 (BI 882370) inhibits the oncogenic BRAF V600E-mutant, the WT BRAF and CRAF kinases with IC50s of 0.4, 0.8, and 0.6 nM, respectively. BI-882370 also inhibits SRC family kinases .
|
-

- HY-112301R
-
|
|
RET
|
Cancer
|
|
Pralsetinib (Standard) is the analytical standard of Pralsetinib. This product is intended for research and analytical applications. Pralsetinib (BLU-667) is a highly potent, selective RET inhibitor. Pralsetinib (BLU-667) inhibits WT RET, RET mutants V804L, V804M, M918T and CCDC6-RET fusion with IC50s of 0.4, 0.3, 0.4, 0.4, and 0.4 nM, respectively .
|
-

- HY-101561
-
|
BLU-285
|
c-Kit
PDGFR
|
Cancer
|
|
Avapritinib (BLU-285) is a highly potent, selective, and orally active KIT and PDGFRA activation loop mutant kinases inhibitor with IC50s of 0.27 and 0.24 nM for KIT D816V and PDGFRA D842V, respectively. Avapritinib (BLU-285) binds the active conformation of the kinase and shows antitumor activity. Avapritinib (BLU-285) attenuates the transport function of both ABCB1 and ABCG2 .
|
-

- HY-131003
-
|
DS-6051b; AB-106; IBI-344
|
ROS Kinase
|
Cancer
|
|
Taletrectinib (DS-6051b) is a potent, orally active, and next-generation selective ROS1/NTRK inhibitor. Taletrectinib potently inhibits recombinant ROS1, NTRK1, NTRK2, and NTRK3 with IC50s of 0.207, 0.622, 2.28, and 0.98 nM, respectively. Taletrectinib also inhibits ROS1 G2032R and other Crizotinib-resistant ROS1 mutants .
|
-

- HY-10357
-
|
|
Organoid
Akt
Autophagy
Apoptosis
|
Cancer
|
|
MK-2206 free base is an orally active, highly potent and selective allosteric Akt inhibitor, with IC50s of 8, 12, and 65 nM for Akt1, Akt2, and Akt3, respectively. Many breast cancer cell lines, and PIK3CA-mutant and cell lines with PTEN loss are sensitive to MK-2206 free base. MK-2206 free base has anticancer activities .
|
-

- HY-153341
-
|
|
PROTACs
Raf
|
Cancer
|
|
CFT1946 is an orally active, CRBN-based and mutant-selective bifunctional degradation activating compound (BiDAC ™) degrader of BRAF V600E with a DC50 of 14 nM in A375 cells. CFT1946 is capable of degrading BRAF V600E (Class I), G469A (Class II), G466V (Class III) mutations, and the p61-BRAF V600E splice variant. CFT1946 can be used in tumor research .
|
-

- HY-14588S2
-
|
ABT-378-d7
|
HIV
SARS-CoV
HIV Protease
Isotope-Labeled Compounds
|
Infection
Cancer
|
|
Lopinavir-d7 is deuterated labeled Lopinavir (HY-14588). Lopinavir (ABT-378) is a highly potent, selective peptidomimetic inhibitor of the HIV-1 protease, with Kis of 1.3 to 3.6 pM for wild-type and mutant HIV protease. Lopinavir acts by arresting maturation of HIV-1 thereby blocking its infectivity . Lopinavir is also a SARS-CoV 3CL pro inhibitor with an IC50 of 14.2 μM .
|
-

- HY-14588R
-
|
ABT-378 (Standard)
|
HIV
HIV Protease
SARS-CoV
|
Infection
Cancer
|
|
Lopinavir (Standard) is the analytical standard of Lopinavir. This product is intended for research and analytical applications. Lopinavir (ABT-378) is a highly potent, selective peptidomimetic inhibitor of the HIV-1 protease, with Kis of 1.3 to 3.6 pM for wild-type and mutant HIV protease. Lopinavir acts by arresting maturation of HIV-1 thereby blocking its infectivity . Lopinavir is also a SARS-CoV 3CL pro inhibitor with an IC50 of 14.2 μM .
|
-

- HY-19719A
-
|
ARQ-092 hydrochloride
|
Akt
Parasite
|
Infection
Cancer
|
|
Miransertib hydrochloride (ARQ-092 hydrochloride) is a potent, orally active, selective and allosteric Akt inhibitor with IC50s of 2.7 nM, 14 nM and 8.1 nM for Akt1, Akt2, Akt3, respectively. Miransertib hydrochloride is also a potent the AKT1-E17K mutant protein inhibitor and has the potential for PI3K/AKT-driven tumors and Proteus syndrome research . Miransertib hydrochloride is effective against Leishmania .
|
-

- HY-144269
-
|
|
Raf
|
Cancer
|
|
SHR902275 is a potent, selective, and orally active RAF inhibitor targeting RAS mutant cancers. SHR902275 has IC50s of 1.6 nM, 10 nM, and 5.7 nM for cRAF, bRAF wt, and bRAF V600E, respectively. SHR902275 shows cell growth inhibition with GI50s of 1.5 and 0.17 nM, 0.4 nM and 0.32 nM for H358, A375, Calu6, and SK-MEL2 cells respectively .
|
-

- HY-19719
-
|
ARQ-092
|
Akt
Parasite
|
Infection
Cancer
|
|
Miransertib (ARQ-092) is a potent, orally active, selective and allosteric Akt inhibitor with IC50s of 2.7 nM, 14 nM and 8.1 nM for Akt1, Akt2, Akt3, respectively. Miransertib is also a potent the AKT1-E17K mutant protein inhibitor and has the potential for PI3K/AKT-driven tumors and Proteus syndrome research . Miransertib is effective against Leishmania .
|
-

- HY-112611
-
|
|
Estrogen Receptor/ERR
|
Cancer
|
|
H3B-5942 is a selective, irreversible and orally active estrogen receptor covalent antagonist, inactivates both wild-type and mutant ERα by targeting Cys530, with Kis of 1 nM and 0.41 nM, respectively. H3B-5942 reduces ERα target gene GREB1, shows potent antitumor activity both in multiple cell lines or animals bearing ERα WT or ERα mutations .
|
-

- HY-172140
-
|
|
Potassium Channel
|
Neurological Disease
|
|
Ebio3 is a selective potassium channel (KCNQ2) inhibitor with an IC50 of 1.2 nM. Ebio3 binds to the KCNQ2 channel through its hydrophobic tail, causing the S6 helix to move inward, which leads to the closure of the inner gate. The inhibitory effect of Ebio3 is also effective in pathogenic mutants of KCNQ2 (such as R75C and I238L), where it can inhibit outward currents by more than 80%. Ebio3 is expected to be used in the research of neurological diseases such as epilepsy .
|
-

- HY-18948
-
GSK321
1 Publications Verification
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
GSK321 is a potent, selective mutant IDH1 inhibitor with IC50 values of 2.9, 3.8, 4.6 and 46 nM for R132G, R132C, R132H and WT IDH1, respectively, and >100-fold selectivity over IDH2. GSK321 induces decrease in intracellular α-Hydroxyglutaric acid (2-HG) (HY-113038B), abrogation of the myeloid differentiation block and induction of granulocytic differentiation at the level of leukemic blasts and more immature stem-like cells. GSK321can be used for research of acute myeloid leukemia (AML) and other cancers .
|
-

- HY-117807
-
|
|
Ras
Farnesyl Transferase
VEGFR
|
Cancer
|
|
A-176120 is a selective inhibitor of farnesyl transferase (IC50=1.2 nM) based on a farnesyl pyrophosphate (FPP) analog, with superior selectivity against GGTaseI (IC50=423 nM), GGTaseII (IC50=3000 nM), and SSase (IC50>10 μM). A-176120 inhibits ras processing in H-ras transformed NIH3T3 cells and HCT116 K-ras mutant cells (ED50=1.6 and 0.5 μM, respectively). A-176120 has antiangiogenic and antitumor activities in vivo and reduces capillary structure formation and VEGF secretion .
|
-

- HY-119396
-
|
|
Endogenous Metabolite
|
Cancer
|
|
DY3002 is a selective and highly potent EGFR inhibitor with activity in overcoming T790M-mediated drug resistance in non-small cell lung cancer. DY3002 exhibited superior inhibitory effects against EGFR T790M mutants in kinase assays (IC50 = 0.71 nM), compared to weaker inhibitory effects against wild-type EGFR (IC50 = 448.7 nM). DY3002 was significantly superior to rociletinib and osimertinib in selectivity, showing an extremely high selectivity index (SI = 632.0). In cell experiments, DY3002 had an IC50 value of 0.037 μM against H1975 cells, showing enhanced inhibitory potency. In addition, DY3002 was superior to other alternative compounds in terms of biological properties and did not cause hyperglycemia .
|
-

- HY-131003A
-
|
DS-6051b free base; AB-106 free base; IBI-344 free base
|
ROS Kinase
|
Cancer
|
|
Taletrectinib (DS-6051b) free base is a potent, orally active, and next-generation selective ROS1/NTRK inhibitor. Taletrectinib free base potently inhibits recombinant ROS1, NTRK1, NTRK2, and NTRK3 with IC50s of 0.207, 0.622, 2.28, and 0.98 nM, respectively. Taletrectinib free base also inhibits ROS1 G2032R and other Crizotinib-resistant ROS1 mutants .
|
-

- HY-122595
-
|
|
Histone Methyltransferase
|
Cancer
|
|
EZH2-IN-1 (compound 3) is a selective and SAM-competitive EZH2 and EZH1 inhibitor with an IC50s of 32 nM, 197 nM and 213 nM for EZH2wt, EZH2 Y641N mutant and EZH1, respectively. EZH2-IN-1 reduces bulk H3K27me3 and H3K27me2 levels. EZH2-IN-1 has the potential for diffuse large B cell lymphoma research .
|
-

- HY-14588S1
-
|
|
Isotope-Labeled Compounds
HIV
HIV Protease
SARS-CoV
|
Infection
|
|
Lopinavir-d8 (ABT-378-d8) is the deuterium labeled Lopinavir. Lopinavir (ABT-378) is a highly potent, selective peptidomimetic inhibitor of the HIV-1 protease, with Kis of 1.3 to 3.6 pM for wild-type and mutant HIV protease. Lopinavir acts by arresting maturation of HIV-1 thereby blocking its infectivity[1][2]. Lopinavir is also a SARS-CoV 3CLpro inhibitor with an IC50 of 14.2 μM[3].
|
-

- HY-135805
-
|
|
EGFR
|
Cancer
|
|
JBJ-04-125-02 is a potent, mutant-selective, allosteric and orally active EGFR inhibitor with an IC50 of 0.26 nM for EGFR L858R/T790M. JBJ-04-125-02 can inhibit cancer cell proliferation and EGFR L858R/T790M/C797S signaling. JBJ-04-125-02 has anti-tumor activities .
|
-

- HY-137458A
-
|
ARQ 751 trihydrochloride
|
Akt
|
Cancer
|
|
Vevorisertib (ARQ 751) trihydrochloride is a selective, allosteric, pan-AKT and AKT1-E17K mutant inhibitors. Vevorisertib trihydrochloride potently inhibit phosphorylation of AKT. Vevorisertib trihydrochloride has Kd values of 1.2 nM and 8.6 nM for AKT1 and AKT1-E17K, respectively. Vevorisertib trihydrochloride has IC50 values of 0.55, 0.81, and 1.3 nM for AKT1, AKT2, and AKT3, respectively. Vevorisertib trihydrochloride can be used for the research of cancer .
|
-

- HY-16936
-
|
|
LRRK2
|
Neurological Disease
|
|
JH-II-127 is an orally active, highly potent, selective and brain-permeable LRRK2 inhibitor, with IC50s of 6, 2 and 48 nM for wild-type LRRK2 and LRRK2-G2019S and mutant LRRK2-A2016T. JH-II-127 inhibits Ser935 phosphorylation in all tissues of mice, including the brain. JH-II-127 can be used in the study of parkinson's syndrome .
|
-

- HY-101561R
-
|
|
c-Kit
PDGFR
|
Cancer
|
|
Avapritinib (Standard) is the analytical standard of Avapritinib. This product is intended for research and analytical applications. Avapritinib (BLU-285) is a highly potent, selective, and orally active KIT and PDGFRA activation loop mutant kinases inhibitor with IC50s of 0.27 and 0.24 nM for KIT D816V and PDGFRA D842V, respectively. Avapritinib (BLU-285) binds the active conformation of the kinase and shows antitumor activity. Avapritinib (BLU-285) attenuates the transport function of both ABCB1 and ABCG2 .
|
-

- HY-128888B
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
(S,R)-GSK321 is the (S,R)-enantiomer of GSK321 (HY-18948). GSK321 is a potent, selective mutant IDH1 inhibitor with IC50 values of 2.9, 3.8, 4.6 and 46 nM for R132G, R132C, R132H and WT IDH1, respectively, and >100-fold selectivity over IDH2. GSK321 induces decrease in intracellular 2-HG, abrogation of the myeloid differentiation block and induction of granulocytic differentiation at the level of leukemic blasts and more immature stem-like cells. GSK321can be used for research of acute myeloid leukemia (AML) and other cancers .
|
-

- HY-114323
-
|
|
PROTACs
FLT3
Apoptosis
STAT
MEK
ERK
|
Cancer
|
|
PROTAC FLT-3 degrader 1 is an effective and selective FLT-3 PROTAC degrader with an IC50 of 0.6 nM. PROTAC FLT-3 degrader 1 inhibits both FLT-3 and FLT-3 ITD mutants. PROTAC FLT-3 degrader 1 has the activity of anti-proliferation and induction of apoptosis, which can be used in the study of tumor. (Pink: FLT3 ligand (HY-168702); Black: Linker (HY-124380); Blue: VHL ligand (HY-125845)) .
|
-

- HY-112289
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
IDH889 is an orally available, brain penetrant, allosteric and mutant specific inhibitor of isocitrate dehydrogenase 1 (IDH1). IDH889 has potent selectivity for IDH1 R132* mutations, with IC50s of 0.02 μM, 0.072 μM and 1.38 μM for IDH1 R132H, IDH1 R132C and IDH1 wt, respectively. IDH889 shows potent cellular inhibition of R-2-hydroxyglutarate (2-HG) production with an IC50 of 0.014 μM .
|
-

- HY-155356
-
|
|
PROTACs
Ras
|
Cancer
|
|
YN14 is a KRASG12C proteolysis targeting chimera (PROTAC). YN14 is highly potent and selective KRASG12C degrader and induces a stable KRASG12C: YN14: VHL ternary complex with low binding free energy (ΔG). YN14 has antiproliferative effects and significantly inhibits KRASG12C-mutant cancer cell growth. YN14 leads to tumor regression with tumor growth inhibition (TGI%) rates more than 100 % in the MIA PaCa-2 xenograft model.
|
-

- HY-14588S
-
|
(rel)-ABT-378-d8
|
Isotope-Labeled Compounds
HIV Protease
SARS-CoV
HIV
|
Others
|
|
(rel)-Lopinavir-d8 ((rel)-ABT-378-d8) is the deuterium labeled Lopinavir (HY-14588) . Lopinavir (ABT-378) is a highly potent, selective peptidomimetic inhibitor of the HIV-1 protease, with Kis of 1.3 to 3.6 pM for wild-type and mutant HIV protease. Lopinavir acts by arresting maturation of HIV-1 thereby blocking its infectivity . Lopinavir is also a SARS-CoV 3CL pro inhibitor with an IC50 of 14.2 μM .
|
-

- HY-159493
-
|
|
Polo-like Kinase (PLK)
|
Cancer
|
|
BI2536-PEG2-Halo is a bifunctional molecule containing a ligand for Halo tag and a Polo-like kinase 1 (PLK1) inhibitor BI-2536 (HY-50698). BI2536-PEG2-Halo inhibits the proliferation of 293T cells with Halo-p53R273H(FL)-mCherry tag (IC50=23 nM), exhibits selective toxicity against p53 mutant cancer cells .
|
-

- HY-136910
-
|
USP7-IN-7
|
Deubiquitinase
MDM-2/p53
|
Cancer
|
|
USP7-797 (USP7-IN-7) is an orally available, selective USP7 inhibitor (IC50=0.5 nmol/L) with antitumor activity. USP7-797 reduces the level of MDM2, thereby increasing the stability and activity of p53, leading to cell cycle arrest and apoptosis. USP7-797 has low nanomolar cytotoxicity against p53 mutant cancer cell lines, p53 wild-type hematological tumors, and neuroblastoma cell lines .
|
-

- HY-100818
-
|
TAS-120
|
FGFR
|
Cancer
|
|
Futibatinib (TAS-120) is an orally bioavailable, highly selective, and irreversible FGFR inhibitor, with IC50s of 3.9, 1.3, 1.6, and 8.3 nM for FGFR 1-4, respectively. Futibatinib inhibits mutant and wild-type FGFR2 with similar IC50s (wild-type FGFR2=0.9 nM; V5651=1-3 nM; N550H=3.6 nM; E566G=2.4 nM) .
|
-

- HY-158101
-
|
CC-94676
|
PROTACs
Androgen Receptor
|
Cancer
|
|
BMS-986365 (CC-94676) is an orally active and selective targeted androgen receptor (AR) PROTAC degrader, capable of inducing cereblon (CRBN) E3 ligase-dependent ubiquitination and degradation of the androgen receptor (AR), as well as various AR mutants. BMS-986365 shows significant in vivo potency, degrading AR, inhibiting AR signaling, and restricting tumor growth in animal models of advanced prostate cancer. (Blue: HY-W247437; Black: linker (HY-W126831); Pink: HY-168697) .
|
-

- HY-150926
-
|
|
Ras
|
Cancer
|
|
G12Si-1 is a selective K-Ras(G12S) covalent inhibitor, which can inhibit oncogenic signaling of K-Ras(G12S). G12Si-1 shows good ability to covalently engage recombinant K-Ras(G12S) at the mutant serine residue. G12Si-1 can also affect nucleotide cycling of K-Ras by blocking Sos-catalyzed exchange and decreasing the rate of EDTA promoted exchange .
|
-

- HY-100818R
-
|
|
FGFR
|
Cancer
|
|
Futibatinib (Standard) is the analytical standard of Futibatinib. This product is intended for research and analytical applications. Futibatinib (TAS-120) is an orally bioavailable, highly selective, and irreversible FGFR inhibitor, with IC50s of 3.9, 1.3, 1.6, and 8.3 nM for FGFR 1-4, respectively. Futibatinib inhibits mutant and wild-type FGFR2 with similar IC50s (wild-type FGFR2=0.9 nM; V5651=1-3 nM; N550H=3.6 nM; E566G=2.4 nM) .
|
-

- HY-152233
-
|
|
HIV
|
Infection
|
|
Reverse transcriptase-IN-4 (compound F10) is a potent and selective non-nucleoside reverse transcriptase (NNRT) inhibitor with an EC50 value of 0.053 μM for wild-type HIV-1 and an EC50 value of 0.26 μM for HIV-1 mutant E138K . Reverse transcriptase-IN-4 is a click chemistry reagent, it contains an Azide group and can undergo copper-catalyzed azide-alkyne cycloaddition reaction (CuAAc) with molecules containing Alkyne groups. It can also undergo strain-promoted alkyne-azide cycloaddition (SPAAC) reactions with molecules containing DBCO or BCN groups.
|
-

- HY-157288
-
|
|
Biochemical Assay Reagents
|
Metabolic Disease
|
|
(R)-Phe-A110/B319, a hapten, is a selective binder to tumor-associated antigens. (R)-Phe-A110/B319 has a 20-fold higher affinity towards the H1047R mutant of p110α in the p110α/p85α PI3K complex. (R)-Phe-A110/B319 can be used for the research of conditional chimeric antigen receptor T (CAR-T) cell activation and tumor targeting .
|
-

- HY-134813A
-
|
|
Ras
|
Cancer
|
|
MRTX1133 formic is a noncovalent, potent, and selective KRAS G12D inhibitor. MRTX1133 formic optimally fills the switch II pocket and extends three substituents to favorably interact with the protein, resulting in an estimated KD against KRAS G12D of 0.2 pM. MRTX1133 formic prevents SOS1-catalyzed nucleotide exchange and/or formation of the KRASG12D/GTP/RAF1 complex, thereby inhibiting mutant KRAS-dependent signal transduction. MRTX1133 formic shows efficacy in tumor models harboring KRAS G12D mutations .
|
-

- HY-159958
-
|
|
Potassium Channel
|
Neurological Disease
|
|
KNa1.1-IN-2 (Compound Z05) is a selective KNa1.1 channel inhibitor with blood-brain barrier penetration, particularly effective against the hERG channel. KNa1.1-IN-2 works by binding to the KNa1.1 channel and blocking the channel activity induced by Gain-of-function (GOF) mutations, effectively intervening in KCNT1-related epilepsy. Additionally, KNa1.1-IN-2 inhibits the GOF mutant Y796H. KNa1.1-IN-2 holds promise for research into KCNT1-related epilepsy disorders .
|
-

- HY-112289B
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
(1R)-IDH889 is the isomer of IDH889 (HY-112289), and can be used as an experimental control. IDH889 is an orally available, brain penetrant, allosteric and mutant specific inhibitor of isocitrate dehydrogenase 1 (IDH1). IDH889 has potent selectivity for IDH1 R132* mutations, with IC50s of 0.02 μM, 0.072 μM and 1.38 μM for IDH1 R132H, IDH1 R132C and IDH1 wt, respectively. IDH889 shows potent cellular inhibition of R-2-hydroxyglutarate (2-HG) production with an IC50 of 0.014 μM .
|
-

- HY-168012
-
|
|
Ras
Phosphatase
|
Cancer
|
|
Pan-RAS-IN-6 (compound 24) is an inhibitor targeting DUSP6, which reduces MAPK activation in the brain of the NCI-H1373-Luc model (DUSP6), at the same time, it shows significant tumor growth inhibition and tumor regression effects in the NSCLC brain metastasis mouse model. Pan-RAS-IN-6 shows high selectivity and strong inhibitory effects, especially in KRAS mutation-related signaling pathways, demonstrating varying inhibitory activity against different KRAS mutants and interacting proteins. The IC50 values for KRAS G12C, G12D, and G12V are 1.3 nM, 4.7 nM, and 0.3 nM, respectively .
|
-

- HY-149508
-
|
|
Keap1-Nrf2
Reactive Oxygen Species
|
Cancer
|
|
Nrf2-IN-3 (Compound R16) is a small-molecule NRF2 inhibitor and increases reactive oxygen species (ROS) production. Nrf2-IN-3 selectively binds KEAP1 mutants and restores their NRF2-inhibitory function by repairing the disrupted KEAP1/NRF2 interactions, leading to proteasome-dependent NRF2 degradation in cells. Nrf2-IN-3 sensitizes KEAP1-mutated tumor cells to Cisplatin (HY-17394), Gefitinib (HY-50895), and KEAP1 G333C-mutated xenograft to Cisplatin .
|
-

- HY-139920
-
|
SH-1028
|
EGFR
|
Cancer
|
|
Oritinib (SH-1028), an irreversible third-generation EGFR TKI, overcomes T790M-mediated resistance in non-small cell lung cancer. Oritinib (SH-1028), a mutant-selective inhibitor of EGFR kinase activity, inhibits EGFR WT, EGFR L858R, EGFR L861Q, EGFR L858R/T790M, EGFR d746-750 and EGFR d746-750/T790M kinases, with IC50s of 18, 0.7, 4, 0.1, 1.4 and 0.89 nM, respectively .
|
-

- HY-136265
-
|
|
Aminoacyl-tRNA Synthetase
|
Cancer
|
|
BC-LI-0186 is a potent and selective inhibitor of Leucyl-tRNA synthetase (LRS; LeuRS) and Ras-related GTP-binding protein D (RagD) interaction (IC50=46.11 nM). BC-LI-0186 competitively binds to the RagD interacting site of LRS (Kd=42.1 nM) and has on effects on LRS-Vps34, LRS-EPRS, RagB-RagD association, mTORC1 complex formation or the activities of 12 kinases. BC-LI-0186 can effectively suppress the activity of cancer-associated?MTOR?mutants and the growth of rapamycin-resistant cancer cells.?BC-LI-0186 is a promising agent for lung cancer research .
|
-

- HY-164476
-
|
|
EGFR
GSK-3
PD-1/PD-L1
|
Cancer
|
|
ES-072 is an orally effective selective EGFR mutant (EGFR-T790M) inhibitor. ES-072 activates GSK3α by inhibiting EGFR-T790M activity, which promotes phosphorylation of PD-L1 at Ser279 and Ser283. The phosphorylated PD-L1 recruits the E3 ubiquitin ligase ARIH1, leading to ubiquitination and proteasomal degradation of PD-L1. This mechanism not only reduces cancer cell growth but also enhances anti-tumor immune response by lowering PD-L1 levels. ES-072 can be used to inhibit proliferation in non-small cell lung cancer (NSCLC) cells .
|
-

- HY-145836
-
|
|
FGFR
|
Cancer
|
|
FGFR4-IN-8 (Compound 7v) is an ATP-competitive, highly selective covalent inhibitor of wild-type and gatekeeper mutant FGFR4. FGFR4-IN-8 exhibits excellent potency against FGFR4, FGFR4 V550L, FGFR4 V550M and FGFR4 C552S with IC50s of 0.5, 0.25, 1.6, 931 nM, respectively. FGFR4-IN-8 exhibits potent antiproliferative activity against Hep3B hepatocellular carcinoma cells with the IC50 value of 29 nM. FGFR4-IN-8 demonstrates modest in vivo antitumor efficacy in nude mice bearing the Huh-7 xenograft model .
|
-

- HY-169920
-
|
|
HIV
Reverse Transcriptase
|
Infection
|
|
HIV-1 inhibitor-79 (Compound 3k) is an HIV inhibitor that exhibits significant inhibitory activity against HIV-1 and its common mutant strains (with IC50 values of 1.9, 1.9, 8.7, and 11 nM against HIV-1, K103, L100I, and E138K, respectively), and has low cytotoxicity and a high selectivity index (CC50 = 21.95 μM, SI = 11478). Additionally, HIV-1 inhibitor-79 also shows antiviral activity against HIV-2, with an EC50 value of 6.14 μM, and significantly inhibits the activity of HIV-1 reverse transcriptase (IC50 = 25 nM) .
|
-

- HY-164552
-
|
|
Apoptosis
Androgen Receptor
|
Cancer
|
|
ZNU-IMB-Z15 (Compound Z15) is an antagonist of the androgen receptor (AR) and also a selective degrader of AR and ARV7. ZNU-IMB-Z15 can directly bind to the ligand-binding domain (LBD) and activation function-1 region of AR, and promote AR degradation through the proteasome pathway. ZNU-IMB-Z15 effectively inhibits the transcriptional activity of AR, AR mutants, and AR splice variants (ARVs), downregulating the mRNA and protein levels of AR downstream target genes, thereby overcoming the resistance to second-generation antiandrogen drugs induced by AR LBD mutations, AR amplification, and ARVs in castration-resistant prostate cancer (CRPC). ZNU-IMB-Z15 can inhibit the proliferation of AR-positive CRPC cell lines and induce their apoptosis, demonstrating anticancer activity both in vivo and in vitro .
|
-

- HY-159922
-
|
|
Androgen Receptor
|
Cancer
|
|
AR antagonist 9 is an orally bioavailable selective androgen receptor (AR) antagonist that exerts anticancer effects by disrupting the dimerization of AR ligand-binding domains, showing potential for overcoming drug resistance in prostate cancer (PCa). Its AR antagonistic activity has an IC50 value of 0.051 μM, comparable to Enzalutamide (HY-70002) (IC50 = 0.060 μM). AR antagonist 9 demonstrated superior efficacy against ARF876L/T877A and ARW741C mutants compared to Enzalutamide (HY-70002). Furthermore, AR antagonist 9 exhibited favorable pharmacokinetic properties, with an oral bioavailability of F = 66.24% in rats. In the LNCaP xenograft mouse model, oral administration of AR antagonist 9 significantly inhibited tumor growth. AR antagonist 9 holds promise for research into overcoming PCa drug resistance .
|
-

- HY-170926
-
|
|
RET
Apoptosis
|
Cancer
|
CQ1373 is a potent RET inhibitor, demonstrating cellular potency with IC50 values of 13.0, 25.7 and 28.4 nM against BaF3 cells expressing CCDC6-RET, CCDC6-RET-G810C and CCDC6-RET-G810R, respectively. CQ1373 exhibits good selectivity toward wild-type RET and solvent front mutants G810C/R with IC50 values of 4.2, 7.1 and 32.4 nM, respectively. CQ1373 inhibits RET phosphorylation and downstream signaling through SHC. CQ1373 induces Apoptosis and cell cycle arrest in BaF3 cells. CQ1373 exhibits anti-tumor efficacy and can be used for cancer research .
|
-

- HY-164392
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
TAS-121 is an orally active, selective, covalent, third-generation mutant EGFR-tyrosine kinase inhibitor (EGFR-TKI). TAS-121 inhibits the L858R mutation (IC50=1.7 nM), Ex19del mutation (IC50=2.7 nM), L858R/T790M mutation (IC50=0.56 nM) and Ex19del/T790M mutation (IC50=1.1 nM) and wild-type EGFR (IC50=8.2 nM). TAS-121 inhibits HER2 and HER4 with IC50s of 110 and 2.6 nM, respectively. TAS-121 inhibits phosphorylation of EGFR and its downstream signaling targets to block cell proliferation. TAS-121 induces apoptosis and displays antitumor activity in SW48 (EGFR G719S) and NCI-H1975 (EGFR L858R/T790M) xenograft models .
|
-

| Cat. No. |
Product Name |
Target |
Research Area |
-
- HY-P3277
-
|
|
Ras
|
Cancer
|
|
KRpep-2d is a potent K-Ras inhibitor and inhibits proliferation of K-Ras-driven cancer cells. KRpep-2d can be used for cancer research .
|
-
- HY-P3277A
-
|
|
Ras
|
Cancer
|
|
KRpep-2d (TFA) is a potent K-Ras inhibitor and inhibits proliferation of K-Ras-driven cancer cells. KRpep-2d can be used for cancer research .
|
| Cat. No. |
Product Name |
Category |
Target |
Chemical Structure |
| Cat. No. |
Product Name |
Chemical Structure |
-
- HY-15772S1
-
|
|
|
Osimertinib- 13C,d3 is the deuterium and 13C labeled Osimertinib. Osimertinib (AZD9291) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M, respectively.
|
-
-
- HY-15772S
-
|
|
|
Osimertinib-d6 is a deuterium labeled osimertinib. Osimertinib is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer[1].
|
-
-
- HY-14588S
-
|
|
|
(rel)-Lopinavir-d8 ((rel)-ABT-378-d8) is the deuterium labeled Lopinavir (HY-14588) . Lopinavir (ABT-378) is a highly potent, selective peptidomimetic inhibitor of the HIV-1 protease, with Kis of 1.3 to 3.6 pM for wild-type and mutant HIV protease. Lopinavir acts by arresting maturation of HIV-1 thereby blocking its infectivity . Lopinavir is also a SARS-CoV 3CL pro inhibitor with an IC50 of 14.2 μM .
|
-
-
- HY-114436S
-
|
|
|
MRTX-1257-d6 is the deuterium labeled MRTX-1257 (HY-114436). MRTX-1257 is a selective, irreversible, covalent and orally active KRAS G12C inhibitor, with an IC50 of 900 pM for KRAS dependent ERK phosphorylation in H358 cells .
|
-
-
- HY-18690S
-
|
|
|
Enasidenib-d6 (AG-221-d6) is the deuterium labeled Enasidenib (HY-18690) . Enasidenib is an oral, potent, reversible, selective inhibitor of the IDH2 mutant enzymes, with IC50s of 100 and 400 nM against IDH2 R140Q and IDH2 R172K, respectively .
|
-
-
- HY-151903S
-
|
|
|
FGFR2/3-IN-1 is a potent and selective FGFR2 and FGFR3 (FGFR) inhibitor with IC50s of 1 nM and 0.5 nM, respectively. FGFR2/3-IN-1 displays >40-fold selectivity over FGFR1/FGFR4 and other kinome. FGFR2/3-IN-1 also inhibits FGFR3 V555L and V555M mutants with IC50s of 2.7 nM and 6.1 nM, respectively[1].
|
-
-
- HY-14588S2
-
|
|
|
Lopinavir-d7 is deuterated labeled Lopinavir (HY-14588). Lopinavir (ABT-378) is a highly potent, selective peptidomimetic inhibitor of the HIV-1 protease, with Kis of 1.3 to 3.6 pM for wild-type and mutant HIV protease. Lopinavir acts by arresting maturation of HIV-1 thereby blocking its infectivity . Lopinavir is also a SARS-CoV 3CL pro inhibitor with an IC50 of 14.2 μM .
|
-
-
- HY-14588S1
-
|
|
|
Lopinavir-d8 (ABT-378-d8) is the deuterium labeled Lopinavir. Lopinavir (ABT-378) is a highly potent, selective peptidomimetic inhibitor of the HIV-1 protease, with Kis of 1.3 to 3.6 pM for wild-type and mutant HIV protease. Lopinavir acts by arresting maturation of HIV-1 thereby blocking its infectivity[1][2]. Lopinavir is also a SARS-CoV 3CLpro inhibitor with an IC50 of 14.2 μM[3].
|
-
| Cat. No. |
Product Name |
|
Classification |
-
- HY-134813
-
|
|
|
Alkynes
|
|
MRTX1133 is a noncovalent, potent, and selective alkyne-based KRAS G12D inhibitor. MRTX1133 optimally fills the switch II pocket and extends three substituents to favorably interact with the protein, resulting in an estimated KD against KRAS G12D of 0.2 pM. MRTX1133 prevents SOS1-catalyzed nucleotide exchange and/or formation of the KRAS G12D/GTP/RAF1 complex, thereby inhibiting mutant KRAS-dependent signal transduction. MRTX1133 selectively inhibits KRAS G12D mutant, but not KRAS wild-type, tumor cells. MRTX1133 has single digit nanomolar activity in cellular assays and marked in vivo efficacy in tumor models harboring KRAS G12D mutations .
|
-
- HY-104066
-
|
Xiliertinib; HMPL-309
|
|
Alkynes
|
|
Theliatinib (Xiliertinib) is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
- HY-159493
-
|
|
|
Azide
|
|
BI2536-PEG2-Halo is a bifunctional molecule containing a ligand for Halo tag and a Polo-like kinase 1 (PLK1) inhibitor BI-2536 (HY-50698). BI2536-PEG2-Halo inhibits the proliferation of 293T cells with Halo-p53R273H(FL)-mCherry tag (IC50=23 nM), exhibits selective toxicity against p53 mutant cancer cells .
|
-
- HY-104066A
-
|
Xiliertinib tartrate; HMPL-309 tartrate
|
|
Alkynes
|
|
Theliatinib (Xiliertinib) tartrate is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib (tartrate) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
- HY-172140
-
|
|
|
Alkynes
|
|
Ebio3 is a selective potassium channel (KCNQ2) inhibitor with an IC50 of 1.2 nM. Ebio3 binds to the KCNQ2 channel through its hydrophobic tail, causing the S6 helix to move inward, which leads to the closure of the inner gate. The inhibitory effect of Ebio3 is also effective in pathogenic mutants of KCNQ2 (such as R75C and I238L), where it can inhibit outward currents by more than 80%. Ebio3 is expected to be used in the research of neurological diseases such as epilepsy .
|
-
- HY-152233
-
|
|
|
Azide
|
|
Reverse transcriptase-IN-4 (compound F10) is a potent and selective non-nucleoside reverse transcriptase (NNRT) inhibitor with an EC50 value of 0.053 μM for wild-type HIV-1 and an EC50 value of 0.26 μM for HIV-1 mutant E138K . Reverse transcriptase-IN-4 is a click chemistry reagent, it contains an Azide group and can undergo copper-catalyzed azide-alkyne cycloaddition reaction (CuAAc) with molecules containing Alkyne groups. It can also undergo strain-promoted alkyne-azide cycloaddition (SPAAC) reactions with molecules containing DBCO or BCN groups.
|
Your information is safe with us. * Required Fields.
Inquiry Information
- Product Name:
- Cat. No.:
- Quantity:
- MCE Japan Authorized Agent:



























































































































































































































