Search Result
Results for "
mutant
" in MedChemExpress (MCE) Product Catalog:
10
Biochemical Assay Reagents
31
Isotope-Labeled Compounds
| Cat. No. |
Product Name |
Target |
Research Areas |
Chemical Structure |
-
- HY-145759
-
|
|
MDM-2/p53
|
Cancer
|
|
Mutant p53 modulator-1 is a mutant p53 modulator. Mutant p53 modulator-1 reduces the progression of cancers that contain a p53 mutation (extracted from patent WO2021231474A1, compound 231B) .
|
-

-
- HY-13984
-
|
|
EGFR
|
Cancer
|
|
Mutant EGFR inhibitor (Example 25) is a potent mutant EGFR inhibitor. Mutant EGFR inhibitor inhibits FGFR WT with an IC50 of 0.41 nM. Mutant EGFR inhibitor inhibits EGFR L858R, EGFR Exon 19 deletion and EGFR T790M. Mutant EGFR inhibitor can be used for the study of diseases mediated by EGFR (particularly cancers) .
|
-
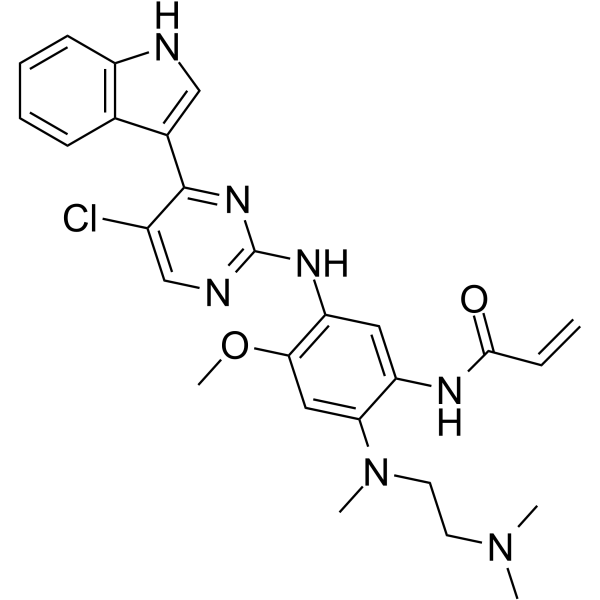
-
- HY-13972
-
-

-
- HY-114459
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Mutant IDH1-IN-4 (compound 434) is an inhibitor of mutant Isocitrate dehydrogenase 1 (IDH 1), with IC50 values of ≤ 0.5 μM for mutant IDH1 in R132H, HT1080 and U87R132H cells .
|
-

-
- HY-18717
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Mutant IDH1-IN-2 is a inhibitor of mutant Isocitrate dehydrogenase (IDH) proteins, with IC50 of in LS-MS biochemical assay, IC50 of 16.6 nM in Fluorescence biochemical assay.
|
-

-
- HY-12475
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Mutant IDH1-IN-1 is a mutant-selective IDH1 inhibitor with with IC50s of 4, 42, 80 and 143 nM against mutant IDH1 R132C/R132C, IDH1 R132H/R132H, IDH1 R132H/WT and wild type IDH1, respectively.
|
-

-
- HY-131312
-
|
LY3410738; mutant IDH1-IN-6
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Mutant IDH1-IN-6 is a potent, selective and orally active mutant isocitrate dehydrogenase (IDH) inhibitor with IC50s of 6.27 nM, 3.71 nM, 36.9 nM and 11.5 nM for IDH1 R132H, IDH1 R132C, IDH2 R140Q and IDH2 R172K mutant enzymes, respectively. Mutant IDH1-IN-6 is less active at inhibiting the IDH wild-type enzymes .
|
-

-
- HY-176916
-
|
|
MDM-2/p53
|
Cancer
|
|
Mutant p53 modulator-2 (Compound 1988) is a mutant p53 modulator. Mutant p53 modulator-2 can be used in the research of cancer .
|
-

-
- HY-100476
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Mutant IDH1-IN-3 (Compound 1) is a selective allosteric inhibitor of mutant isocitrate dehydrogenase 1 (IDH1). Mutant IDH1-IN-3 has an IC50 of 13 nM for R132H IDH1. Mutant IDH1-IN-3 inhibits the production of D-2-hydroxyglutaric acid (2HG) in cells. Mutant IDH1-IN-3 can be used in the study of cancer .
|
-

-
- HY-158319
-
|
|
Apoptosis
Isocitrate Dehydrogenase (IDH)
NAMPT
|
Cancer
|
|
Mutant IDH1/NAMPT-IN-1 (Compound 23h) is a dual inhibitor of mutant isocitrate dehydrogenase 1 (mutant IDH1) (IC50=14.93 nM) and nicotinamide phosphoribosyltransferase (NAMPT) (IC50=12.56 nM). Mutant IDH1/NAMPT-IN-1 can induce apoptosis. Mutant IDH1/NAMPT-IN-1 crosses the blood-brain barrier effectively .
|
-

-
- HY-175213
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Mutant IDH1-IN-7 is a highly selective Isocitrate Dehydrogenase 1 (IDH1) R132H (IC50 = 0.26 μM, Kd = 2.1 μM)/R132C (IC50 = 1.1 μM) inhibitor. Mutant IDH1-IN-7 has no inhibitory effect on wild-type IDH1, IDH2-wt, and IDH2 R140Q. Mutant IDH1-IN-7 inhibits 2-Hydroxyglutarate (2-HG) production in U87-MG R132H cells (EC50 = 0.55 μM). Mutant IDH1-IN-7 exhibits moderate antiproliferative effects on U87-MG R132H and HT-1080 cells .
|
-

-
- HY-131312A
-
|
LY3410738 (gentisate); mutant IDH1-IN-6 (gentisate)
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Crelosidenib (gentisate) is a potent, selective and orally active mutant isocitrate dehydrogenase (IDH) inhibitor with IC50 of 6.27 nM, 3.71 nM, 36.9 nM and 11.5 nM for IDH1 R132H, IDH1 R132C, IDH2 R140Q and IDH2 R172K mutant enzymes, respectively. Crelosidenib (gentisate) is less active at inhibiting the IDH wild-type enzymes .
|
-

-
- HY-155005
-
|
|
EGFR
|
Cancer
|
|
EGFR mutant-IN-2 (Compound D51) is an EGFR mutant inhibitor. EGFR mutant-IN-2 inhibits the EGFR L858R/T790M/C797S mutant with an IC50 value of 14 nM. EGFR mutant-IN-2 inhibits the EGFR del19/T790M/C797S mutant with an IC50 value of 62 nM. EGFR mutant-IN-2 has favorable PK parameters, safety properties, in vivo stability, and antitumor activity .
|
-

-
- HY-132920
-
|
|
Ras
|
Cancer
|
|
KRAS mutant protein inhibitor 1 is a KRAS mutant protein inhibitor for potential research in cancer.
|
-

-
- HY-125841
-
|
|
EGFR
|
Cancer
|
|
EGFR mutant-IN-1, a 5-methylpyrimidopyridone derivative, is a potent and selective EGFR L858R/T790M/C797S mutant inhibitor with an IC50 of 27.5 nM, while being a significantly less potent for EGFR WT (IC50 >1.0 μM) .
|
-

-
- HY-E70880
-
|
EndoCC N180H
|
Glycosidase
|
Others
|
|
Endoglycosidase CC (N180H mutant) (EndoCC N180H) is a mutant endoglycosidase, which efficiently and specifically recognizes core fucose and O-GlcNAc .
|
-

-
- HY-126298
-
|
|
Raf
|
Cancer
|
|
RAF mutant-IN-1 is a RAF kinase inhibitor, extracted from patent WO2019107987A1, with IC50 values of 21 nM, 30 nM and 392 nM for C-RAF 340D/Y341D, B-RAF V600E and B-RAF WT, respectively .
|
-

-
- HY-E70881
-
|
EndoF3 D165A
|
Glycosidase
|
Others
|
|
Endoglycosidase F3 (D165A mutant) (EndoF3 D165A) is a mutant endoglycosidase, which efficiently and specifically recognizes core fucose and O-GlcNAc .
|
-

-
- HY-E70882
-
|
EndoD N322Q
|
Glycosidase
|
Others
|
|
Endoglycosidase D (N322Q mutant) (EndoD N322Q) is a mutant endoglycosidase, which can be highly efficient to transfer Man5GlcNAc oxazoline to a similar cyclic glycopeptide carrying two free GlcNAc moieties to give a doubly glycosylated peptide .
|
-

-
- HY-E70884
-
|
EndoS2 D184M
|
Glycosidase
|
Others
|
|
Endoglycosidase S2 (D184M mutant) (EndoS2 D184M) is a mutant endoglycosidase that facilitates the transfer of a glycan-oxazoline donor with a defined glycoform to the Fc region of IgG antibodies .
|
-

-
- HY-E70883
-
|
EndoS D233Q
|
Glycosidase
|
Others
|
|
Endoglycosidase S (D233Q mutant) (EndoS D233Q) is a mutant endoglycosidase, which catalyzes the glycosylation of Trastuzumab (HY-P9907)-GlcNAc with the functionalized, non-natural glycans to give glycosylated monoclonal Abs (mAbs) carrying two glycans each functionalized with two reaction handles .
|
-

-
- HY-E70879
-
|
EndoM N175Q
|
Glycosidase
|
Others
|
|
Endoglycosidase M (N175Q mutant) (EndoM N175Q) can transfer natural N-glycans or oxazoline N-glycans to any peptide or protein with a GlcNAc residue to form a β1-4-glycosidic linkage. Endoglycosidase M (N175Q mutant) is a useful tool in the synthesis of homogeneous glycopeptides and glycoproteins .
|
-

-
- HY-118731
-
|
|
Reverse Transcriptase
|
Infection
|
|
TNK-6123 is a potent Emivirine (HY-15353) analogue with improved activity against drug-resistant HIV mutants. TNK-6123 improves activity against Lys103Asn mutant RT .
|
-

-
- HY-D1056C5
-
|
LPS, from Salmonella enterica (Serotype minnesota Re 595 (Re mutant))
|
Toll-like Receptor (TLR)
Bacterial
|
Inflammation/Immunology
|
Lipopolysaccharides (LPS), from S. enterica (Salmonella enterica) serotype minnesota Re 595 (Re mutant) is prepared from Salmonella enterica strain Re 595 (Re mutant). The structure in the LPS of strain Re 595 was shown to induce secretion and aggregation in human platelets .
It is recommended to prepare a solution with concentration ≥2 mg/mL. Vortex thoroughly for more than 10 minutes. Due to the adsorption characteristics of LPS, silanized container or low adsorption centrifuge tubes should be used for aliquoting and storage, and mix thoroughly before use.
|
-

-
- HY-123636
-
|
|
p97
|
Cancer
|
|
UPCDC-30245 is an allosteric p97 inhibitor with an IC50 of approximately 27 nM . UPCDC-30245 inhibits the p97 mutant N660K similar to wild type (WT; IC50=300 nM) and shows 3-fold resistance for p97 mutant T688A . UPCDC-30245 can be used in the research of cancer .
|
-

-
- HY-P2361
-
|
|
Ras
|
Others
|
|
S12 is a mutant RAS peptide containing the Gly (G) to Ser (S12) substitution. The sequence of the peptide is KLVVVGASGVGKS .
|
-

-
- HY-168580
-
|
|
c-Kit
|
Cancer
|
|
c-Kit-IN-7 (Compound 104) is a potent inhibitor of c-Kit, with IC50 values of ≤10 nM. c-Kit-IN-7 inhibits the cell proliferation in GIST430 and BaF3 mutants, with IC50 values of ≤100 nM. c-Kit-IN-7 plays an important role in cancer driven by c-KIT kinase mutations research .
|
-

-
- HY-176354S
-
-

-
- HY-176358S
-
-

-
- HY-176342S
-
-

-
- HY-176359S
-
-

-
- HY-176353S
-
-

-
- HY-176357S
-
-

-
- HY-176341S
-
-

-
- HY-108636
-
|
|
MDM-2/p53
|
Cancer
|
|
RETRA is a mutant p53-dependent activator of p73 that suppresses mutant p53-bearing cancer cells. RETRA increases the expression level of p73, and a release of p73 from the blocking complex with mutant p53, which produces tumor-suppressor effects .
|
-

-
- HY-141566
-
|
|
Histone Methyltransferase
|
Cancer
|
|
EZH2-IN-5 is a potent EZH2 inhibitor with IC50 values of 1.52 nM and 4.07 nM for wild-type and mutant Tyr641 EZH2, respectively .
|
-

-
- HY-153767
-
|
|
MDM-2/p53
|
Cancer
|
|
PK095 is a p53 mutant stabilizer. PK095 can be used for research of cancer .
|
-

-
- HY-150927
-
|
|
Ras
|
Cancer
|
|
G12Si-2, an analog of G12Si-1 (HY-150926), is a negative control tool. G12Si-2 is not a covalent inhibitor of the G12S mutant of K-Ras .
|
-

-
- HY-131067
-
|
|
EGFR
|
Cancer
|
|
EMI56, the derivative of EMI1, displays greater potency toward mutant EGFR than EMI1. EMI56 inhibits EGFR triple mutants .
|
-

-
- HY-153974
-
|
|
EGFR
|
Cancer
|
|
BBT-176 is an orally effective EGFR inhibitor. BBT-176 has inhibitory activity against multiple EGFR C797S mutant cell lines. BBT-176 is commonly used in cancer research .
|
-

-
- HY-P2670
-
|
|
NF-κB
|
Others
|
|
SN50M, a mutant peptide of SN50 (HY-P0151), is a cell membrane-permeable inactive control peptide .
|
-

-
- HY-156819
-
|
RMC-9805; KRAS G12D inhibitor 18
|
Ras
Apoptosis
|
Cancer
|
|
Zoldonrasib (RMC-9805) is a potent and orally active KRAS G12D inhibitor.Zoldonrasib induces apoptosis in KRAS G12D mutant cancer cells. Zoldonrasib has the potential for the research of KRAS G12D mutant cancer .
|
-

-
- HY-114226
-
|
FT-2102
|
Isocitrate Dehydrogenase (IDH)
|
Inflammation/Immunology
Cancer
|
|
Olutasidenib (FT-2102) is a highly potent, orally active, brain penetrant and selective inhibitor of mutant Isocitrate dehydrogenase 1 (IDH1), with IC50 values of 21.2 nM and 114 nM for IDH1- R132H and IDH1- R132C, respectively . Olutasidenib (FT-2102) is under the study in the treatment of acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS) .
|
-

-
- HY-110144
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
TC-E 5008 is a potent mutant IDH1 inhibitor with Ki values of 190 nM and 120 nM for R132H and R132C IDH1 mutants, respectively. TC-E 5008 exhibits very weak activity against WT-IDH1 with a Ki value of 12.3 μM. TC-E 5008 has anti-proliferative activity on multiple ER positive breast cancer cell lines .
|
-

-
- HY-P2360
-
|
Ras 5-17
|
Ras
|
Others
|
|
G12 (Ras 5-17) is a wild-type Ras peptide consisted of amino acids 5-17 (KLVVVGAGGVGKS). G12 can be used as a control of mutant Ras peptides studies (such V12) .
|
-

-
- HY-160449
-
|
|
MDM-2/p53
|
Cancer
|
|
p53 Activator 10 (Example C-2) is a compound that targets the y220c mutant of p53. p53 Activator 10 activation is involved in the downstream effects of tumor suppression .
|
-

-
- HY-P2250
-
|
ELA-32 negative control
|
Apelin Receptor (APJ)
|
Others
|
|
ELA RR>GG (ELA-32 negative control), an ELABELA (ELA-32 human) mutant peptide, is inactive. ELA RR>GG is a negative control for ELABELA (HY-P2196) .
|
-

-
- HY-15352
-
|
DPC 083
|
Reverse Transcriptase
HIV
|
Infection
|
|
BMS 561390 (DPC 083) is an orally available non-nucleoside reverse transcriptase inhibitor (NNRTI) with broad inhibitory effects on wild-type HIV-1 and mutant strains .
|
-

-
- HY-P2360A
-
|
Ras 5-17 TFA
|
Ras
|
Others
|
|
G12 (Ras 5-17) TFA is a wild-type Ras peptide consisted of amino acids 5-17 (KLVVVGAGGVGKS). G12 TFA can be used as a control of mutant Ras peptides studies (such V12) .
|
-

-
- HY-149464
-
|
|
Ras
|
Cancer
|
|
ARN22089 is a oral active novel class of trisubstituted pyrimidine, blocks the interaction of CDC42 GTPases with specific downstream effectors. ARN22089 blocks tumor growth in BRAF mutant mouse melanoma model .
|
-

- HY-172919
-
|
|
Phosphodiesterase (PDE)
NAMPT
Apoptosis
|
Cancer
|
|
PDEδ/NAMPT IN-1 (Compound 17d) is a dual inhibitor targeting phosphodiesterase 6 (PDE6) (KD=0.410 nM) and nicotinamide phosphoribosyl transferase (NAMPT) (IC50=2.21 nM). PDEδ/NAMPT IN-1 blocks KRAS-related signal transduction and interferes with the synthesis of nicotinamide adenine dinucleotide (NAD +), inducing apoptosis in KRAS mutant pancreatic cancer cells. PDEδ/NAMPT IN-1 is promising for research of KRAS mutant pancreatic cancer .
|
-

- HY-164542
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
TQ05310 is an orally available inhibitor of IDH2 mutants, targeting both IDH2-R140Q (IC50=136.9 nM) and IDH2-R172K (IC50=37.9 nM) mutants. TQ05310 inhibits the production of 2-hydroxyglutarate (2-HG) and induces differentiation of cells expressing IDH2-R140Q and IDH2-R172K by inhibiting the enzymatic activity of mutant IDH2. TQ05310 can be used for the study of acute myeloid leukemia .
|
-

- HY-118169
-
|
|
Bacterial
|
Infection
|
|
A-Factor is an inducer of streptomycin biosynthesis in an inactive mutant of Streptomyces griseus. In addition, A-Factor can also induce spore formation during conidial development of Magnaporthe oryzae .
|
-

- HY-153724
-
|
|
Ras
|
Cancer
|
|
BI-2865 is a none-covalent pan-KRAS Inhibitor. BI-2865 binds to WT, G12C, G12D, G12V and G13D mutant KRAS with KDs of 6.9, 4.5, 32, 26, 4.3 nM respectively. BI-2865 inhibits the proliferation of G12C, G12D or G12V mutant KRAS expressing BaF3 cells (mean IC50: roughly 140 nM) .
|
-

- HY-126047
-
|
|
Biochemical Assay Reagents
|
Others
|
|
(S)-(-)-Anatabine is a Ti plasmid mutant.
(S)-(-)-Anatabine has the activity of inducing tobacco blastoma to transform
into nicotinic alkaloids. (S)-(-)-Anatabine can be used in the study of
biotransformation of cotyl teratoma .
|
-

- HY-145594
-
|
AB-291; DS-1001
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Safusidenib (AB-291; DS-1001) is an orally bioavailable, selective mutant IDH1 inhibitor. Safusidenib strongly inhibits mutant IDH1 but not wild-type IDH1. Safusidenib impairs tumor activity in chondrosarcoma . Safusidenib exhibits activity against IDH1R132H, and IDH1R132C with IC50s of 15, and 130 nM in assays without any preincubation, respectively .
|
-

- HY-106918
-
|
R86183; TIBO R 86183
|
Reverse Transcriptase
HIV
|
Infection
|
|
Tivirapine (R86183) is a nonnucleoside HIV-1 RT inhibitor against HIV-1-induced cytopathic effects with an EC50 value of 4 nM. Tivirapine inhibits the Yl8lC mutant of HIV-1 RT .
|
-

- HY-129545
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
DS-1001b is an orally active, blood-brain permeable, potent IDH-1 (isocitrate dehydrogenase-1) mutant inhibitor. DS-1001b has antitumor activity .
|
-

- HY-124761
-
|
|
Polo-like Kinase (PLK)
Autophagy
Mitosis
|
Cancer
|
|
Poloppin is a potent, cell penetrant inhibitor of the mitotic Polo-like kinase (PLK) (IC50=26.9 μM) and prevents the protein-protein interaction via the Polo-box domain (PBD) (Kd= 29.5 μM). Poloppin selectively kills cells expressing mutant KRAS, enhancing death in mitosis. Poloppin is used for the study of KRAS-mutant cancers as single agents, or in combination with c-MET inhibitors .
|
-

- HY-19634
-
|
|
Sirtuin
|
Cancer
|
|
YK-3-237, a SIRT1 activator, targets mutant p53. YK-3-237 inhibits the proliferation of triple-negative breast cancer cells .
|
-

- HY-150905
-
|
|
EGFR
|
Cancer
|
|
EGFR ligand-2 (compound C4), a covalent EGFR ligand, is a EGFR mutant inhibitor with IC50s of 21 nM and 48 nM for EGFR L858R and EGFR L858R/T790M, respectively. EGFR ligand-2 can be used to synthesize PROTAC .
|
-

- HY-145022
-
|
|
Ras
|
Cancer
|
|
KRAS G12C inhibitor 25 is a KRAS G12C inhibitor. KRAS G12C inhibitor 25 inhibits SOSl-assisted GDP/GTP exchanging activity of KRAS-G12C mutant (IC50=0.48 nM). From WO2021216770A1 compound 3 .
|
-

- HY-126905
-
|
NSC328784
|
Reactive Oxygen Species (ROS)
|
Cancer
|
|
ZMC3 (NSC328784) is a zinc chelator. ZMC3 shows Zinc metallochaperone properties. ZMC3 shows enhanced sensitivity in p53-R175H mutant cells. ZMC3 increases the cell ROS level .
|
-

- HY-131909
-
|
|
Anaplastic lymphoma kinase (ALK)
|
Cancer
|
|
CJ-2360 is a potent and orally active ALK inhibitor with IC50s of 2.2, 4.0, 8.8, 6.3, and 8.9 nM against wild-type ALK and F1197M, G1269A, L1196M, and S1206Y ALK mutants, respectively. CJ-2360 displays potent inhibitory activity against two clinically reported ALK mutants (C1156Y and L1196M) and a few other kinases (LTK, MERTK, CLK1, DAPK1, and DAPK2) among the 468 kinases evaluated .
|
-

- HY-P10425
-
|
|
Histone Methyltransferase
|
Cancer
|
|
T2857W is a mutant peptide that has inhibitory effect on KIX-MLL interaction (IC50=5.67 μM). T2857W can be used for protein-protein interaction (PPI) and for the study of diseases related to KIX-MLL interaction, such as leukemia, cancer, etc .
|
-

- HY-108887
-
|
|
Raf
|
Cancer
|
|
Anticancer agent 124 is an orally active, highly selective and potent pan RAF inhibitor. Anticancer agent 124 inhibits MAPK signalling in BRAF V600E, NRAS and KRAS mutant tumor cells .
|
-

- HY-141640
-
|
|
PROTACs
EGFR
|
Cancer
|
|
MS154 is a first-in-class E3 ligase cereblon-recruited EGFR degrader with Kd values of 1.8 nM and 3.8 nM for EGFR WT and EGFR L858R mutant, respectively. MS154 potently induces degradation of mutant, but not wild-type, EGFR in cancer cell lines in an E3 ligase-dependent manner. MS154 exhibits anticancer effects against lung cancer (blue: E3 ligase ligand (HY-103596); black: linker (HY-W096167); pink: EGFR ligand (HY-168305)) .
|
-

- HY-157458
-
|
|
ATTECs
Phosphodiesterase (PDE)
Autophagy
|
Cancer
|
|
PDEδ autophagic degrader 1 (compound 12c), an autophagosome-tethering compound (ATTEC). is a potent PDEδ autophagic degrader. PDEδ autophagic degrader 1 reduces the PDEδ protein level through lysosome-mediated autophagy without affecting the PDEδ mRNA expression. PDEδ autophagic degrader 1 suppresses the growth in KRAS mutant pancreatic cancer cells .
|
-

- HY-14588
-
|
ABT-378
|
HIV
HIV Protease
SARS-CoV
|
Infection
Cancer
|
|
Lopinavir (ABT-378) is a highly potent, selective peptidomimetic inhibitor of the HIV-1 protease, with Kis of 1.3 to 3.6 pM for wild-type and mutant HIV protease. Lopinavir acts by arresting maturation of HIV-1 thereby blocking its infectivity . Lopinavir is also a SARS-CoV 3CL pro inhibitor with an IC50 of 14.2 μM .
|
-

- HY-123390
-
|
|
Bcr-Abl
Akt
|
Cancer
|
|
DB07107 is a potent agent resistant T315I mutant Bcr-Abl tyrosine kinase inhibitor. DB07107 is also a potent Akt1 inhibitor with an IC50 value of 360 nM .
|
-

- HY-19617A
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-1 hydrochloride is an orally active and irreversible L858R/T790M mutant selective EGFR inhibitor. EGFR-IN-1 hydrochloride potently inhibits Gefitinib-resistant EGFR L858R, T790M with 100-fold selectivity over wild-type EGFR. EGFR-IN-1 hydrochloride displays strong antiproliferative activity against the H1975 cells and the first line mutant HCC827 cells. Antitumor activity .
|
-

- HY-159493
-
|
|
Polo-like Kinase (PLK)
|
Cancer
|
|
BI2536-PEG2-Halo is a bifunctional molecule containing a ligand for Halo tag and a Polo-like kinase 1 (PLK1) inhibitor BI-2536 (HY-50698). BI2536-PEG2-Halo inhibits the proliferation of 293T cells with Halo-p53R273H(FL)-mCherry tag (IC50=23 nM), exhibits selective toxicity against p53 mutant cancer cells .
|
-

- HY-19706
-
ARS-853
4 Publications Verification
|
Ras
Apoptosis
|
Cancer
|
|
ARS-853 is a cell-active, selective, covalent KRAS G12C inhibitor with an IC50 of 2.5 μM. ARS-853 inhibits mutant KRAS-driven signaling by binding to the GDP-bound oncoprotein and preventing activation .
|
-

- HY-164524
-
|
|
Eukaryotic Initiation Factor (eIF)
Akt
NF-κB
|
Cancer
|
|
SBI-0640726 is an eIF4G1 inhibitor with antiproliferative activity in melanoma. SBI-0640726 disrupts the eIF4F translation initiation complex by inhibiting AKT and NF-kB signaling pathways. SBI-0640726 inhibits the growth of NRAS and BRAF mutant melanoma in vitro .
|
-

- HY-128888
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
(R,R)-GSK321 is a wild-type isocitrate dehydrogenase 1 (WT IDH1) inhibitor with an IC50 value of 120 nM. (R,R)-GSK321 as a mutant R132H IDH1 inhibitor, is an isomer of GSK321 with some wild-type cross reactivity .
|
-

- HY-143319
-
|
|
EGFR
|
Cancer
|
|
SPH5030 is a selective and irreversible HER2 inhibitor. SPH5030 inhibits HER2 WT and EGFR WT with IC50s of 3.51 and 8.13 nM, respectively. SPH5030 shows excellent activities against HER2 mutants. SPH5030 can be used for the research of cancer .
|
-

- HY-19617B
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-1 TFA is an orally active and irreversible L858R/T790M mutant selective EGFR inhibitor. EGFR-IN-1 TFA potently inhibits Gefitinib-resistant EGFR L858R, T790M with 100-fold selectivity over wild-type EGFR. EGFR-IN-1 TFA displays strong antiproliferative activity against the H1975 cells and the first line mutant HCC827 cells. Antitumor activity .
|
-

- HY-19617
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-1 (compound 24) is an orally active and irreversible L858R/T790M mutant selective EGFR inhibitor. EGFR-IN-1 potently inhibits Gefitinib-resistant EGFR L858R, T790M with 100-fold selectivity over wild-type EGFR. EGFR-IN-1 displays strong antiproliferative activity against the H1975 cells and the first line mutant HCC827 cells. Antitumor activity .
|
-

- HY-161923
-
|
|
EGFR
Apoptosis
Akt
ERK
|
Cancer
|
|
EGFR-IN-120 (Compound 11eg) is an orally active EGFR inhibitor. EGFR-IN-120 inhibits EGFR L858R/T790M/C797S with an IC50 value of 0.053 μM, and has a relatively weak effect on EGFR WT (IC50: 1.05 μM). EGFR-IN-120 inhibits the phosphorylation of EGFR and main downstream effectors (STAT3, AKT, and Erk). EGFR-IN-120 induces cell cycle arrest and cell apoptosis in EGFR mutant cells. EGFR-IN-120 inhibits the proliferation of the NSCLC cells harboring EGFR L858R/T790M/C797S with an IC50 of 0.052 μM .
|
-

- HY-14588S2
-
|
ABT-378-d7
|
HIV
SARS-CoV
HIV Protease
Isotope-Labeled Compounds
|
Infection
Cancer
|
|
Lopinavir-d7 is deuterated labeled Lopinavir (HY-14588). Lopinavir (ABT-378) is a highly potent, selective peptidomimetic inhibitor of the HIV-1 protease, with Kis of 1.3 to 3.6 pM for wild-type and mutant HIV protease. Lopinavir acts by arresting maturation of HIV-1 thereby blocking its infectivity . Lopinavir is also a SARS-CoV 3CL pro inhibitor with an IC50 of 14.2 μM .
|
-

- HY-142160
-
|
|
Raf
|
Cancer
|
|
GNE-9815 (compound 7) is a highly selective, pan-RAF inhibitor with good oral bioavailability. GNE-9815 exhibits Ki values of 0.062 and 0.19 nM for CRAF and BRAF, respectively. GNE-9815 combines with MEK inhibitor Cobimetinib (HY-13064) shows synergistic modulation of MAPK pathway. GNE-9815 can be used in studies of KRAS mutant cancers .
|
-

- HY-155114
-
|
|
HIV
|
Infection
|
|
HIV-1 inhibitor-59 (Compound I-5b) is a HIV-1 inhibitor, with EC50s of 5.62-171 nM against the wild-type (WT) and mutant HIV-1 strains. HIV-1 inhibitor-59 has moderate RT enzyme inhibitory activity (IC50: 0.094-12.0 μM) .
|
-

- HY-101489
-
|
|
PDGFR
Bcr-Abl
Apoptosis
|
Cancer
|
|
GZD856 formic is a potent and orally active PDGFRα/β inhibitor, with IC50s of 68.6 and 136.6 nM, respectively. GZD856 formic is also a Bcr-Abl T315I inhibitor, with IC50s of 19.9 and 15.4 nM for native Bcr-Abl and the T315I mutant. GZD856 formic has antitumor activity .
|
-

- HY-171499
-
|
EL625
|
MDM-2/p53
|
Cancer
|
|
Cenersen (EL625) is an oligonucleotide targeting TP53. Cenersen can eliminate the activity of TP53 gain-of-function mutants and increase the sensitivity of lymphoma cells to cytotoxic chemotherapy in vitro. Cenersen can be used for the study of chronic lymphocytic leukemia (CLL) .
|
-

- HY-138072
-
|
|
EGFR
|
Cancer
|
|
EMI1 is an EGFR ex19del/T790M/C797S and EGFR L858R/T790M/C797S inhibitor. EMI1 can be used for the research of mutant EGFR-associated, drug-resistant non-small-cell lung cancer (NSCLC) .
|
-

- HY-151287
-
|
|
Ras
|
Cancer
|
|
KRAS inhibitor-20 is a small molecular inhibitor of KRas G12C, the oncogenic mutant. KRAS inhibitor-20 inhibits KRas G12C with the IC50 value <10 nM .
|
-

- HY-143234
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
mIDH1-IN-1 (compound 43) is a potent and selective mIDH1 (mutant isocitrate dehydrogenases 1) inhibitor, with an IC50 of 961.5 nM. mIDH1-IN-1 potently inhibits intracellular 2-HG (2-hydroxyglutarate) production in HT1080 cells, with an EC50 of 208.6 ± 8.0 nM. mIDH1-IN-1 shows a significant anti-proliferation activity on IDH1 mutant-U-87 cells, with an IC50 of 41.8 nM. mIDH1-IN-1 is an antitumor agent, and can be used for IDH1 mutated solid tumors research .
|
-

- HY-P5082
-
|
|
α-synuclein
|
Neurological Disease
|
|
α-Synuclein 4554W is an inhibitor of α-Synuclein (aSyn) aggregation with associated toxicity. α-Synuclein 4554W consists of GIVNGVKA sequences, previously identified through intracellular library screening. α-Synuclein 4554W reduces fibril formation of aSyn mutants assocaited with Parkinson’s disease .
|
-

- HY-177511
-
|
|
Ras
p38 MAPK
Raf
MEK
ERK
|
Cancer
|
|
KRAS G12D-IN-30 (Compound 4) is a KRAS inhibitor. KRAS G12D-IN-30 inhibits the activation of the downstream MAPK signaling cascade (Raf1-MEK-ERK) by blocking the activity of the KRAS G12 mutant. KRAS G12D-IN-30 can be used for the research of cancer .
|
-

- HY-108232
-
MK-2206
Maximum Cited Publications
419 Publications Verification
|
Organoid
Akt
Autophagy
Apoptosis
|
Cancer
|
|
MK-2206 is an orally active, highly potent and selective allosteric Akt inhibitor, with IC50s of 8, 12, and 65 nM for Akt1, Akt2, and Akt3, respectively. Many breast cancer cell lines, and PIK3CA-mutant and cell lines with PTEN loss are sensitive to MK-2206. MK-2206 has anticancer activities .
|
-

- HY-148070
-
|
|
FLT3
FAK
Cytochrome P450
|
Cancer
|
|
FLT3-IN-17 inhibits CYPs and FLT3 mutants activity (IC50s: <0.5 nM for D835Y). FLT3-IN-17 is also a FAK inhibitor, with an IC50 value of 12 nM. FLT3 ligand-2 can be used in the research of cancers .
|
-

- HY-122914
-
|
|
Ras
|
Cancer
|
|
KRAS inhibitor-3 is an inhibitor of KRAS inhibitor. KRAS inhibitor-3 binds to WT and oncogenic KRAS mutants with high affinity (KD: 0.28 μM for KRAS WT, 0.63 μM for KRAS G12C, 0.37 μM for KRAS G12D, 0.74 μM for KRAS Q61H). KRAS inhibitor-3 also disrupts interaction of KRAS with Raf .
|
-

- HY-10357
-
|
|
Organoid
Akt
Autophagy
Apoptosis
|
Cancer
|
|
MK-2206 free base is an orally active, highly potent and selective allosteric Akt inhibitor, with IC50s of 8, 12, and 65 nM for Akt1, Akt2, and Akt3, respectively. Many breast cancer cell lines, and PIK3CA-mutant and cell lines with PTEN loss are sensitive to MK-2206 free base. MK-2206 free base has anticancer activities .
|
-

- HY-165559
-
|
|
TRP Channel
|
Inflammation/Immunology
|
|
Trpvicin is a potent and subtype-selective TRPV3 inhibitor with IC50s of 0.41 and 0.22 μM for hTRPV3-WT and hTRPV3-G573S mutant, respectively. Trpvicin exhibits minimal effects on other TRP family members (such as TRPV1/2/4/5/6, TRPA1 and TRPM8). Trpvicin inhibits the TRPV3 channel by stabilizing it in a closed state via VSLD-PD binding. Trpvicin accesses additional binding sites inside the central cavity of the G573S mutant to remodel symmetry and block the channel. Trpvicin inhibits itch and hair loss in mouse models. Trpvicin can be used for study of inflammation and immunology .
|
-

- HY-136789
-
|
BDTX-189
|
EGFR
|
Cancer
|
|
Tuxobertinib (BDTX-189) is a potent, orally active and selective inhibitor of allosteric EGFR and HER2 oncogenic mutations, including EGFR/HER2 exon 20 insertion mutants. Tuxobertinib shows KDs of 0.2, 0.76, 13 and 1.2 nM for EGFR, HER2, BLK and RIPK2, reapectively. Anticancer activity .
|
-

- HY-115282A
-
|
TRC-253
|
Androgen Receptor
|
Cancer
|
|
JNJ-63576253 (TRC-253) is a potent and orally active full antagonist of androgen receptor (AR), with IC50s of 37 and 54 nM for F877L mutant AR and wild-type AR in LNCaP cells. JNJ-63576253 can be used for the research of castration-resistant prostate cancer (CRPC) .
|
-

- HY-168514
-
|
|
SOS1
EGFR
|
Cancer
|
|
SOS1/EGFR-IN-2 (Compound 4) is a SOS1 and EGFR dual inhibitor with IC50s of 8.3 and 14.6 nM, respectively. SOS1/EGFR-IN-2 exhibits significant antiproliferative effect on the cancer cells haboring various KRAS mutants .
|
-

- HY-101489A
-
|
|
PDGFR
Bcr-Abl
Apoptosis
|
Cancer
|
|
GZD856 formic is a potent and orally active PDGFRα/β inhibitor, with IC50s of 68.6 and 136.6 nM, respectively. GZD856 formic is also a Bcr-Abl T315I inhibitor, with IC50s of 19.9 and 15.4 nM for native Bcr-Abl and the T315I mutant. GZD856 formic has antitumor activity . GZD856 (formic) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-152560
-
|
|
HIV
|
Infection
|
|
HIV-1 inhibitor-55 (compound 4d) inhibits WT HIV-1 with an EC50 value of 8.6 nM. HIV-1 inhibitor-55 also shows inhibitory potency against single and double HIV-1 mutants. HIV-1 inhibitor-55 can be used for the research of virus infection .
|
-

- HY-143306
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
IDH1 Inhibitor 5 (compound 2) is an IDH1 (isocitrate dehydrogenase 1) inhibitor. IDH1 Inhibitor 5 inhibits MOG cells and wild-type IDH1 glioma cells with expressing exogenous mutant IDH1 R132H protein with IC50s of 64.4 and 34.9 nM, respectively .
|
-

- HY-15519
-
|
|
IAP
FLT3
|
Cancer
|
|
LBW242, a 3-mer and Smac mimetic, is a potent and orally active proapoptotic IAP inhibitor. LBW242 shows effects on mutant FLT3-expressing cells. LBW242 has activity against multiple myeloma, and potentiates TRAIL- and anticancer agent-mediated cell death of ovarian cancer cells .
|
-

- HY-115282
-
|
TRC-253 free base
|
Androgen Receptor
|
Cancer
|
|
JNJ-63576253 (TRC-253) free base is a potent and orally active full antagonist of androgen receptor (AR), with IC50s of 37 and 54 nM for F877L mutant AR and wild-type AR in LNCaP cells. JNJ-63576253 free base can be used for the research of castration-resistant prostate cancer (CRPC) .
|
-

- HY-130616
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
EGFR-IN-11 is a fourth-generation EGFR-tyrosine kinase inhibitor (EGFR-TKI) with an IC50 of 18 nM for triple mutant EGFR L858R/T790M/C797S. EGFR-IN-11 significantly suppresses the EGFR phosphorylation, induce the apoptosis, and arrest cell cycle at G0/G1 .
|
-

- HY-122209
-
|
|
HBV
|
Infection
|
|
DVR-01 is a HBV inhibitor with EC50 values of 1.7 and 1.6 μM in AML12HBV10 and HepDES19 cells, respectively. DVR-01 shows antiviral activity against drug-resistant HBV mutants with EC50s of 2.403-3.273 μM. DVR-01 can be used for the research of HBV infection and related diseases .
|
-

- HY-167135
-
|
|
HIV
Reverse Transcriptase
|
Infection
|
|
DPC-082 is a potent non-nucleoside reverse transcriptase inhibitor (NNRTI) of HIV-1. DPC-082 inhibits wild-type RF virus with an IC90 of 2.0 nM. DPC-082 inhibits various single and many multiple amino acid substituted HIV-1 mutant viruses .
|
-

- HY-W004705
-
|
2-Hydroxymethylaniline
|
Chloride Channel
|
Endocrinology
|
|
(2-Aminophenyl)methanol (2-Hydroxymethylaniline) is a molecular chaperone to rescue P123S mutant pendrin. (2-Aminophenyl)methanol has the advantages of low dose, long-term effect and low toxicity. (2-Aminophenyl)methanol can be used for the study of Pendred syndrome (a syndromic deafness) .
|
-

- HY-156158
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
IDH2 R140Q-IN-2 (compound 36) is an an orally active IDH2 R140Q inhibitor (IC50: 29 nM). IDH2 R140Q-IN-2 reduces D2HG production in TF-1 cell lines expressing mutant IDH2 R140Q (IC50: 10 nM). IDH2 R140Q-IN-2 suppresses D2HG levels in tumor tissue. IDH2 R140Q-IN-2 can be used for research of acute myeloid leukemia (AML) .
|
-

- HY-16271
-
|
4-Isothioureidobutyronitrile hydrochloride; thioureidobutyronitrile hydrochloride; thioureido butyronitrile hydrochloride
|
MDM-2/p53
Apoptosis
|
Cancer
|
|
Kevetrin hydrochloride is a potent activator of p53, induces apoptosis in TP53 wild-type and mutant acute myeloid leukemia cells. Kevetrin a preferential cytotoxic activity against blast cells .
|
-

- HY-161654
-
|
|
SOS1
PROTACs
Ras
|
Cancer
|
|
PROTAC SOS1 degrader-10 (Compound 11o) is a degrader for son of sevenless 1 (SOS1) in a CRBN and proteasome dependent manner. PROTAC SOS1 degrader-10 degrades SOS1 in KRAS mutant cancer cells SW620, A549 and DLD-1, with DC50s of 2.23, 1.85 and 7.53 nM, respectively. PROTAC SOS1 degrader-10 inhibits the proliferations of cells SW620, A549 and DLD-1, with IC50s of 36.7, 52.2 and 107 nM, respectively. PROTAC SOS1 degrader-10 inhibits phosphorylation of ERK. (Pink: SOS1 ligand (HY-161655); Black: linker (HY-161656); Blue: E3 ligase ligand (HY-W249500))
|
-

- HY-111836
-
|
|
Molecular Glues
β-catenin
|
Cancer
|
|
NRX-252114 is a potent enhancer of the interaction between β-catenin, and its cognate E3 ligase, SCF β-TrCP. NRX-252114 enhances the binding of pSer33/S37A β-catenin peptide for β-TrCP with an EC50 of 6.5 nM and a Kd of 0.4 nM. NRX-252114 induces mutant β-catenin degradation .
|
-

- HY-122862
-
|
|
Ras
|
Cancer
|
|
RAS inhibitor Abd-7, a potent RAS-binding compound (Kd=51 nM), is a RAS-effector protein-protein interaction (PPI) inhibitor. RAS inhibitor Abd-7 interacts with RAS inside the cells, prevents RAS-effector interactions and inhibits endogenous RAS-dependent signaling. RAS inhibitor Abd-7 impairs the PPI of various mutant KRAS proteins with PI3K, CRAF and RALGDS as well as NRAS Q61H and HRAS G12V .
|
-
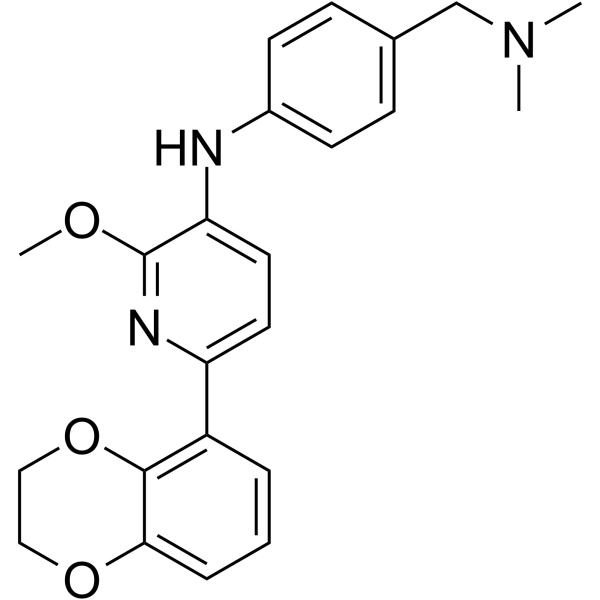
- HY-134813
-
|
|
Ras
|
Cancer
|
|
MRTX1133 is a noncovalent, potent, and selective alkyne-based KRAS G12D inhibitor. MRTX1133 optimally fills the switch II pocket and extends three substituents to favorably interact with the protein, resulting in an estimated KD against KRAS G12D of 0.2 pM. MRTX1133 prevents SOS1-catalyzed nucleotide exchange and/or formation of the KRAS G12D/GTP/RAF1 complex, thereby inhibiting mutant KRAS-dependent signal transduction. MRTX1133 selectively inhibits KRAS G12D mutant, but not KRAS wild-type, tumor cells. MRTX1133 has single digit nanomolar activity in cellular assays and marked in vivo efficacy in tumor models harboring KRAS G12D mutations .
|
-

- HY-10574
-
|
R278474; TMC278; DB08864
|
HIV
Reverse Transcriptase
|
Infection
|
|
Rilpivirine (R278474) is a potent and specific diarylpyrimidine (DAPY) non-nucleoside reverse transcriptase inhibitor (NNRTI). Rilpivirine has high antiviral activity against wild-type HIV (EC50=0.4 nM) and mutant viruses (EC50=0.1-2.0 nM). Rilpivirine has a high genetic barrier to resistance development of HIV .
|
-

- HY-10574A
-
|
TMC-278 hydrochloride; TMC278 hydrochloride; TMC 278 hydrochloride
|
SARS-CoV
MMP
|
Infection
|
|
Rilpivirine (R278474) hydrochloride is a potent and specific diarylpyrimidine (DAPY) non-nucleoside reverse transcriptase inhibitor (NNRTI). Rilpivirine hydrochloride has high antiviral activity against wild-type HIV (EC50=0.4 nM) and mutant viruses (EC50=0.1-2.0 nM). Rilpivirine hydrochloride has a high genetic barrier to resistance development of HIV .
|
-

- HY-174851
-
|
|
Ras
|
Cancer
|
|
KRAS G12C-IN-70 is a selective KRAS G12C mutant inhibitor. KRAS G12C-IN-70 blocks KRAS G12C-mediated downstream signaling pathways (e.g., RAF-MEK-ERK) and inhibits tumor cell proliferation. KRAS G12C-IN-70 is promising for research of KRAS G12C mutation-related tumors (such as non-small cell lung cancer, colorectal cancer) .
|
-

- HY-10624
-
THIQ
1 Publications Verification
|
Melanocortin Receptor
|
Metabolic Disease
|
|
THIQ is the first selective agonist of the melanocortin-4 receptor (MC4R), with high affinity and potency for hMC4R (IC50=1.2 nM, EC50=2.1 nM) and rMC4R (IC50=0.6 nM, EC50=2.9 nM). THIQ maintains low potency at MC1R, MC3R and MC5R. THIQ plays a role in eliciting erectile activity in rodents. THIQ acts as a pharmacoperone of the MC4R rescuing the cell surface expression and signaling of some intracellularly retained MC4R mutants .
|
-

- HY-156529
-
|
|
Ras
|
Cancer
|
|
pan-KRAS-IN-2 (Compound 6) is a pan-inhibitor with IC50s ≤ 10 nM for KRAS WT and mutants (G12D, G12C, G12V, G12S, G12A and Q61H); and an IC50 > 10 μM for KRAS G13D. pan-KRAS-IN-2 can be used to study various KRAS-mediated cancers, such as pancreatic cancer and colorectal cancer .
|
-

- HY-150754
-
|
|
Bacterial
|
Infection
|
|
FtsZ-IN-4 is an orally active FtsZ (filamenting temperature-sensitive mutant Z) inhibitor, exhibits excellent antibacterial activity. FtsZ-IN-4 shows good pharmaceutical properties with low cytotoxicity (CC50 >20 μg/mL) .
|
-

- HY-149880
-
|
|
c-Met/HGFR
|
Cancer
|
|
c-Met-IN-18 is ATP competitive type-III c-MET inhibitor of WT and D1228V mutant c-MET. c-Met-IN-18 has inhibitory for WT/D1228V with an IC50 value of 0.013/0.20 e.c-Met-IN-18 can be used for the research of c-MET driven cancers . c-Met-IN-18 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-134813A
-
|
|
Ras
|
Cancer
|
|
MRTX1133 formic is a noncovalent, potent, and selective KRAS G12D inhibitor. MRTX1133 formic optimally fills the switch II pocket and extends three substituents to favorably interact with the protein, resulting in an estimated KD against KRAS G12D of 0.2 pM. MRTX1133 formic prevents SOS1-catalyzed nucleotide exchange and/or formation of the KRASG12D/GTP/RAF1 complex, thereby inhibiting mutant KRAS-dependent signal transduction. MRTX1133 formic shows efficacy in tumor models harboring KRAS G12D mutations .
|
-

- HY-10574AR
-
|
TMC-278 hydrochloride (Standard); TMC278 hydrochloride (Standard); TMC 278 hydrochloride (Standard)
|
Reference Standards
SARS-CoV
MMP
|
Infection
|
|
Rilpivirine (hydrochloride) (Standard) is the analytical standard of Rilpivirine (hydrochloride). This product is intended for research and analytical applications. Rilpivirine (R278474) hydrochloride is a potent and specific diarylpyrimidine (DAPY) non-nucleoside reverse transcriptase inhibitor (NNRTI). Rilpivirine hydrochloride has high antiviral activity against wild-type HIV (EC50=0.4 nM) and mutant viruses (EC50=0.1-2.0 nM). Rilpivirine hydrochloride has a high genetic barrier to resistance development of HIV .
|
-

- HY-10574R
-
|
R278474 (Standard); TMC278 (Standard); DB08864 (Standard)
|
Reference Standards
HIV
Reverse Transcriptase
|
Infection
|
|
Rilpivirine (Standard) is the analytical standard of Rilpivirine. This product is intended for research and analytical applications. Rilpivirine (R278474) is a potent and specific diarylpyrimidine (DAPY) non-nucleoside reverse transcriptase inhibitor (NNRTI). Rilpivirine has high antiviral activity against wild-type HIV (EC50=0.4 nM) and mutant viruses (EC50=0.1-2.0 nM). Rilpivirine has a high genetic barrier to resistance development of HIV .
|
-

- HY-114491A
-
|
|
ERK
Raf
|
Cancer
|
|
Rineterkib hydrochloride (compound B) is an orally available ERK1 and ERK2 inhibitor in the treatment of a proliferative disease characterized by activating mutations in the MAPK pathway. The activity is particularly related to the treatment of KRAS-mutant NSCLC, BRAF-mutant NSCLC, KRAS-mutant pancreatic cancer, KRAS-mutant colorectal cancer (CRC) and KRAS-mutant ovarian cancer. Rineterkib hydrochloride can also inhibit RAF .
|
-

- HY-114491
-
|
|
ERK
Raf
|
Cancer
|
|
Rineterkib (compound B) is an orally available ERK1 and ERK2 inhibitor in the treatment of a proliferative disease characterized by activating mutations in the MAPK pathway. The activity is particularly related to the treatment of KRAS-mutant NSCLC, BRAF-mutant NSCLC, KRAS-mutant pancreatic cancer, KRAS-mutant colorectal cancer (CRC) and KRAS-mutant ovarian cancer. Rineterkib hydrochloride can also inhibit RAF .
|
-

- HY-12083
-
|
|
Bcr-Abl
|
Cancer
|
|
PPY-A is a potent T315I mutant and wild-type Abl kinases inhibitor with IC50s of 9 and 20 nM, respectively. PPY-A inhibits Ba?F3 cells transformed with wild-type Abl and Abl T315I mutantl with IC50s of 390 and 180 nM, respectively. PPY-A can be used for the research of chronic myeloid leukemia (CML) .
|
-

- HY-124089
-
|
|
Cannabinoid Receptor
|
Metabolic Disease
|
|
Eicosapentaenoyl ethanolamide, an omega-3 fatty acid, is one of N-acylethanolamines (NAEs). Eicosapentaenoyl ethanolamide is cannabinoid CB1/CB2 receptor agonist. Eicosapentaenoyl ethanolamide acts as a metabolic signal. Eicosapentaenoyl ethanolamide inhibits dietary restriction (DR)-induced lifespan extension in wild type animals and suppresses lifespan extension in a TOR pathway mutant .
|
-

- HY-P2265
-
|
|
SOS1
Ras
|
Cancer
|
|
SAH-SOS1A is a peptide-based SOS1/KRAS protein interaction inhibitor. SAH-SOS1A binds to wild-type and mutant KRAS (G12D, G12V, G12C, G12S, and Q61H) with nanomolar affinity (EC50=106-175 nM), directly and independently blocks nucleotide association, impairs KRAS-driven cancer cell viability, and exerts its effects by on-mechanism blockade of the ERK-MAPK phosphosignaling cascade downstream of KRAS .
|
-

- HY-169536
-
|
WAY-326769
|
CFTR
|
Inflammation/Immunology
|
|
CFTR activator 2 (WAY-326769) is an activator of mutant cystic fibrosis transmembrane conductance regulator (mutant-CFTR) .
|
-

- HY-179219S
-
|
|
DNA/RNA Synthesis
|
Cancer
|
|
RTx-303 is an orally active, selective DNA polymerase θ (Polθ) inhibitor (IC50 = 5.1 nM). RTx-303 exhibits significantly high cellular potency and strongly potentiates PARPi in BRCA1/2 mutant cells and patient-derived xenograft models. RTx-303 can be used for the study of BRCA2-mutated breast cancer .
|
-

- HY-153220
-
|
|
PROTACs
Btk
|
Cancer
|
|
NX-2127 (Compound 28) is an orally active PROTAC deggrader, targeting to Bruton’s Tyrosine Kinase (Btk) . NX-2127 inhibits proliferation of BTK C481S mutant TMD8 cells, more effectively than Ibrutinib (HY-10997). NX-2127 catalyzes the degradation of Ikaros (IKZF1) and Aiolos (IKZF3) with of 25 nM and 54 nM, respectively. NX-2127 stimulates T cell activation and increases IL-2 production in primary human T Cells . (Pink: BTK ligand 10 (HY-168302); Black: (R)-4-(1-(Pyrrolidin-3-ylmethyl)piperidin-4-yl)aniline (HY-168348); Blue: Thalidomide 5-fluoride (HY-W087383)
|
-

- HY-175864
-
|
|
EGFR
Apoptosis
p38 MAPK
ERK
Akt
STAT
|
Inflammation/Immunology
Cancer
|
|
EGFR-IN-173 is an orally active, pan-mutant EGFR tyrosine kinase inhibitor that targets EGFR 19del, L858R/T790M and C797S triple-mutations, potently inhibiting EGFR 19del/T790M/C797S with an IC50 of 1.19 nM while showing over 100-fold selectivity for mutant over wild-type EGFR (IC50 = 19.362 μM against WT). EGFR-IN-173 significantly inhibits cell migration, induces apoptosis in non-small cell lung cancer (NSCLC) cells. EGFR-IN-173 inhibits EGFR phosphorylation and suppresses the downstream pathways (MAPK/ERK, AKT, STAT3). EGFR-IN-173 exhibits antitumor efficacy in NSCLC and Ba/F3 xenograft models. EGFR-IN-173 can be used for NSCLC research .
|
-

- HY-145120
-
|
RG6344; RO7276389; B-Raf IN 2
|
Raf
|
Cancer
|
|
BRAF inhibitor. RG6344 can be used for the study of BRAF V600-mutant solid tumors, such as colorectal cancer (CRC) .
|
-

- HY-P2265A
-
|
|
SOS1
Ras
|
Cancer
|
|
SAH-SOS1A TFA is a peptide-based SOS1/KRAS protein interaction inhibitor. SAH-SOS1A TFA binds to wild-type and mutant KRAS (G12D, G12V, G12C, G12S, and Q61H) with nanomolar affinity (EC50=106-175 nM). SAH-SOS1A TFA directly and independently blocks nucleotide association. SAH-SOS1A TFA impairs KRAS-driven cancer cell viability and exerts its effects by on-mechanism blockade of the ERK-MAPK phosphosignaling cascade downstream of KRAS .
|
-

- HY-19985
-
|
|
EGFR
|
Cancer
|
|
PF-06459988 is an orally activity, irreversible and mutant-selective inhibitor of EGFR mutant forms. PF-06459988 demonstrates high potency and specificity to the T790M-containing double mutant EGFRs. PF-06459988 can be used for the research of cancer .
|
-

- HY-155046
-
|
|
FGFR
|
Cancer
|
|
FGFR4-IN-14 (Compound 27i) is a FGFR4 inhibitor (IC50: 2.4 nM. FGFR4-IN-14 inhibits the proliferation of V550L and N535K mutant strains, with IC50s of 21 nM, 2.5 nM, 171 nM against huh7, BaF3/ETV6-FGFR4-V550L and BaF3/ETV6-FGFR4-N535K cells respectively. FGFR4-IN-14 has potent antitumor efficacy in the Huh7 xenograft model. FGFR4-IN-14 can be used for research of hepatocellular carcinoma (HCC) .
|
-

- HY-12972
-
|
PF-06747775
|
EGFR
|
Cancer
|
|
Mavelertinib is a selective, orally available and irreversible EGFR tyrosine kinase inhibitor (EGFR TKI), with IC50s of 5, 4, 12 and 3 nM for Del, L858R, and double mutants T790M/L858R and T790M/Del, respectively. Mavelertinib can be used for the research of non-small-cell lung cancer (NSCLC) .
|
-

- HY-19803
-
|
ASP8273 mesylate
|
EGFR
|
Cancer
|
|
Naquotinib mesylate (ASP8273 mesylate) is an orally available, mutant-selective and irreversible EGFR inhibitor; with IC50s of 8-33 nM toward EGFR mutants and 230 nM for EGFR.
|
-

- HY-19729
-
|
ASP8273
|
EGFR
|
Cancer
|
|
Naquotinib (ASP8273) is an orally available, mutant-selective and irreversible EGFR inhibitor; with IC50s of 8-33 nM toward EGFR mutants and 230 nM for EGFR.
|
-

- HY-159795
-
|
PF-07799933; ARRY-440
|
Raf
PERK
|
Cancer
|
|
Claturafenib ( PF-07799933) is an orally active inhibitor of pan-mutant BRAF. Claturafenib inhibits pERK in cells (IC50 value of 1.6 nM in HT29 cells). Claturafenib has anticancer activity against BRAF G469A mutant NSCLC and BRAF K601E mutant melanoma .
|
-

- HY-131066
-
|
|
EGFR
|
Cancer
|
|
EMI48, the derivative of EMI1, displays greater potency toward mutant EGFR than EMI1. EMI48 inhibits EGFR triple mutants .
|
-

- HY-18948
-
GSK321
1 Publications Verification
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
GSK321 is a potent, selective mutant IDH1 inhibitor with IC50 values of 2.9, 3.8, 4.6 and 46 nM for R132G, R132C, R132H and WT IDH1, respectively, and >100-fold selectivity over IDH2. GSK321 induces decrease in intracellular α-Hydroxyglutaric acid (2-HG) (HY-113038B), abrogation of the myeloid differentiation block and induction of granulocytic differentiation at the level of leukemic blasts and more immature stem-like cells. GSK321can be used for research of acute myeloid leukemia (AML) and other cancers .
|
-

- HY-U00447
-
|
|
MDM-2/p53
DNA Alkylator/Crosslinker
|
Cancer
|
|
PK11000 is an alkylating agent, and stabilizes the DNA-binding domain of both WT and mutant p53 proteins by covalent cysteine modification without compromising DNA binding. PK11000 has anti-tumor activities .
|
-

- HY-122595
-
|
|
Histone Methyltransferase
|
Cancer
|
|
EZH2-IN-1 (compound 3) is a selective and SAM-competitive EZH2 and EZH1 inhibitor with an IC50s of 32 nM, 197 nM and 213 nM for EZH2wt, EZH2 Y641N mutant and EZH1, respectively. EZH2-IN-1 reduces bulk H3K27me3 and H3K27me2 levels. EZH2-IN-1 has the potential for diffuse large B cell lymphoma research .
|
-

- HY-164467
-
|
|
Wnt
|
Cancer
|
|
CCT070535 blocks TCF-dependent transcription at the TCF level, with GI50 values of 17.6 μM, 11.1 μM, 11.8 μM and 13.4 μM in HT29 (APC mutant), HCT116 (oncogenic β-catenin), SW480 (APC mutant) and SNU475 (Axin mutant) cells, respectively .
|
-

- HY-19980A
-
PRIMA-1
2 Publications Verification
NSC-281668
|
Autophagy
MDM-2/p53
Ferroptosis
Apoptosis
|
Cancer
|
|
PRIMA-1 (NSC-281668) is a mutant p53 reactivator, restores the sensitivity of TP53 mutant-type thyroid cancer cells to the histone methylation inhibitor 3-Deazaneplanocin A.
|
-

- HY-12872
-
|
EGF816
|
EGFR
|
Cancer
|
|
Nazartinib (EGF816) is a covalent mutant-selective EGFR inhibitor, with Ki and Kinact of 31 nM and 0.222 min −1 on EGFR(L858R/790M) mutant, respectively.
|
-

- HY-158151
-
|
|
MDM-2/p53
|
Cancer
|
|
p53 Activator 12 (compound 510B) is a potent p53 activator. p53 Activator 12 binds to mutant p53 and restores the ability of the p53 mutant to bind DNA .
|
-

- HY-12872A
-
|
EGF816 mesylate
|
EGFR
|
Cancer
|
|
Nazartinib mesylate (EGF816 mesylate) is a novel, covalent mutant-selective EGFR inhibitor, with Ki and Kinact of 31 nM and 0.222 min −1 on EGFR(L858R/790M) mutant, respectively.
|
-

- HY-142645
-
|
|
RSV
|
Infection
|
|
TP0591816 is a potent dual inhibitor of wild-type and mutant respiratory syncytial virus fusion proteins (wild-type, EC50 = 0.27 nM; D486N-mutant, EC50 = 0.70 nM).
|
-

- HY-145836
-
|
|
FGFR
|
Cancer
|
|
FGFR4-IN-8 (Compound 7v) is an ATP-competitive, highly selective covalent inhibitor of wild-type and gatekeeper mutant FGFR4. FGFR4-IN-8 exhibits excellent potency against FGFR4, FGFR4 V550L, FGFR4 V550M and FGFR4 C552S with IC50s of 0.5, 0.25, 1.6, 931 nM, respectively. FGFR4-IN-8 exhibits potent antiproliferative activity against Hep3B hepatocellular carcinoma cells with the IC50 value of 29 nM. FGFR4-IN-8 demonstrates modest in vivo antitumor efficacy in nude mice bearing the Huh-7 xenograft model .
|
-

- HY-149359
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
IHMT-IDH1-053 (compound 16) is a highly selectivity and irreversible IDH1-mutant inhibitor with an IC50 of 4.7 nM for IDH1 R132H. IHMT-IDH1-053 displays high selectivity against IDH1 mutants over IDH1 wt and IDH2 wt/mutants. IHMT-IDH1-053 inhibits 2-hydroxyglutarate (2-HG) production in IDH1 R132H mutant transfected 293T cells (IC50=28 nM). IHMT-IDH1-053 binds to the IDH1 R132H protein in the allosteric pocket adjacent to the NAPDH binding pocket through a covalent bond with residue Cys269. IHMT-IDH1-053 inhibits the proliferation of HT1080 cell line and primary AML cells which both bear IDH1 R132 mutants .
|
-

- HY-W034419
-
|
|
MDM-2/p53
Apoptosis
|
Cancer
|
|
STIMA-1 can stimulate mutant p53 DNA binding in vitro and induce expression of p53 target proteins and trigger apoptosis in mutant p53-expressing human tumor cells .
|
-

- HY-144104
-
-

- HY-128860
-
|
|
EGFR
|
Cancer
|
|
Mutated EGFR-IN-2 (compound 91) is a mutant-selective EGFR inhibitor extracted from patent WO2017036263A1, which potently inhibits single-mutant EGFR (T790M) and double-mutant EGFR (including L858R/T790M (IC50=<1nM) and ex19del/T790M), and can suppress activity of single gain-of-function mutant EGFR (including L858R and ex19del) as well. Mutated EGFR-IN-2 shows anti-tumor antivity .
|
-

- HY-121708
-
|
|
c-Kit
|
Cancer
|
|
KI-328 is a novel inhibitor targeting KIT kinase that has selective activity against some KIT mutant kinases commonly found in acute myeloid leukemia (AML). KI-328 showed specificity for KIT kinase in in vitro kinase assays and inhibited the growth of wild-type (Wt) and mutant KIT-expressing cells, but had lower activity against D816V-KIT. Comparative analysis of the inhibitory effects of several potent KIT inhibitors on the growth of multiple mutant KIT-expressing cells showed that the multi-kinase inhibitors had comparable activity against D816V-KIT as against other mutant KITs; however, heat shock protein 90 (HSP90) inhibitors showed significant activity against D816V-KIT, inhibiting the growth of D816V-KIT-expressing cells at concentrations that did not affect the growth of other mutant KIT-expressing cells. These results suggest that potent KIT inhibitors have different activities against different types of KIT mutant kinases. Therefore, in clinical development, KIT inhibitors need to validate their activity against multiple types of KIT mutant kinases.
|
-

- HY-19540
-
GSK864
5 Publications Verification
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
GSK864, a chemical probe, is an isocitrate dehydrogenase 1 (IDH1) mutant inhibitor; inhibits IDH1 mutants R132C, R132H, and R132G with IC50 values of 8.8, 15.2 and 16.6 nM.
|
-

- HY-19726
-
|
|
MDM-2/p53
|
Cancer
|
|
NSC59984 induces mutant p53 protein degradation via MDM2 and the ubiquitin-proteasome pathway . NSC59984 acts by targeting GOF-mutant p53 and stimulates p73 to restore the p53 pathway signaling .
|
-

- HY-113843
-
|
|
MDM-2/p53
|
Cancer
|
|
RETRA (hydrobromide) is a mutant p53-dependent activator of p73 that can inhibit cancer cells carrying mutant p53. RETRA (hydrobromide) increases the expression level of p73, induces transcriptional activation of several common to transcriptional targets p53 and p73, which leads to mutant p53- and p73-dependent inhibition of tumor growth, reduction of colony formation and induction of effector caspases .
|
-

- HY-158115
-
|
|
Molecular Glues
Raf
MEK
|
Cancer
|
|
NST-628 is a brain-permeable MAPK pathway molecule glue that inhibits RAF phosphorylation and MEK activation. NST-628 also binds RAF and prevents the formation of BRAF-CRAF and BRAF-ARAF heterodimers, effectively inhibiting the RAS-MAPK pathway. NST-628 inhibits RAS- and RAF-driven cancers and demonstrated potent inhibition in mutant KRAS, NRAS, BRAF class II/III, and NF1-mutant tumors .
|
-

- HY-156671
-
|
|
Ras
PI3K
ERK
mTOR
Apoptosis
|
Cancer
|
|
RMC-4998 is an orally active inhibitor targeting the active or GTP-bound state of the KRAS G12C mutant. RMC-4998 can form a ternary complex with intracellular CYPA and the activated KRAS G12C mutant, with an IC50 value of 28 nM. RMC-4998 can inhibit ERK signaling in KRAS G12C mutant cancer cells and induce apoptosis. RMC-4998 can be used for tumor research .
|
-

- HY-W423595
-
|
|
EGFR
|
Cancer
|
|
BEBT-109 is a potent pan-mutant-selective EGFR inhibitor. BEBT-109 has improved pharmacokinetic properties. BEBT-109 can be used for multiple mutant-EGFR-driven non-small cell lung cancer (NSCLC) research .
|
-

- HY-120331
-
|
|
HIV
HIV Protease
|
Infection
|
|
U-89360E is an inhibitor for HIV-1 protease. U-89360E inhibits the protease of HIV-1 wildtype, V82D mutant and V82N mutant with Ki of 20 nM, 560 nM and 2100 nM, respectively .
|
-

- HY-160287
-
|
|
Epigenetic Reader Domain
|
Cancer
|
|
NVS MLLT-1 (compound 3), a chemical probe, is a potent and selective MLLT1 inhibitor with Kd values of 0.18 μM, 0.24 μM, and 0.22 μM for wild-type, T1 mutants, and T3 mutants, respectively .
|
-

- HY-W007626
-
|
|
Fungal
|
Infection
|
|
3,5-Dimethoxybenzaldehyde is an antifungal agent. 3,5-Dimethoxybenzaldehyde exhibits antifungal activity against Saccharomyces cerevisiae cell wall integrity mutants (slt2Δ and bck1Δ) and Aspergillus fumigatus MAPK mutants (sakAΔ and mpkCΔ) .
|
-

- HY-145020
-
|
|
Ras
|
Cancer
|
|
KRAS G12C inhibitor 23 is a KRAS G12C inhibitor. KRAS G12C inhibitor 23 inhibits H358 cells with an IC50 of 491 nM (WO2021218939A1, compound 1) .
|
-

- HY-114636
-
|
|
Insecticide
|
Others
|
|
RG-102240 is a ligand for insect ecdysone receptor (EcR), with IC50 of 85 and 13 nM, in wildtype G:CfE(DEF) and its A110P mutant. RG-102240 induces the reporter gene activity of wild-type CfEcR and A110P mutant .
|
-

- HY-151523
-
|
|
Ras
|
Cancer
|
|
KRas G12R inhibitor 1 is a covalent inhibitor targeting the common oncogenic mutant KRas G12R with selectivity for the mutant arginine. KRas G12R inhibitor 1 possesses an α,β-diketoamide and exploits strong nucleophilicity of the mutant cysteine and irreversibly binds in the Switch II region of KRas. KRas G12R inhibitor 1 can be researched for K-Ras (G12R)-driven cancer such as pancreatic ductal adenocarcinoma (PDAC) .
|
-

- HY-112802A
-
|
|
c-Kit
|
Cancer
|
|
AZD3229 Tosylate is a potent pan-KIT mutant inhibitor for the treatment of gastrointestinal stromal tumors.
|
-

- HY-100520
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-2 is a a noncovalent, irreversible, mutant-selective second generation EGFR inhibitor.
|
-

- HY-176870
-
|
|
Ras
|
Cancer
|
|
KRAS-IN-42 (Compound Z1063) is a covalent KRAS G12D mutants inhibitor. KRAS-IN-42 is promising for research of KRAS G12D-mutant cancers (e.g., non-small cell lung cancer, colorectal cancer) .
|
-

- HY-156681A
-
|
|
PI3K
|
Cancer
|
|
(S)-STX-478 is the S-enantiomer of STX-478. STX-478 is a selective inhibitor of PI3Kα mutants, preventing metabolic dysfunction and demonstrating antitumor activity in xenograft mouse models with PI3Kα mutant tumors .
|
-

- HY-100601
-
|
|
MDM-2/p53
|
|
|
PK7242 is an inducer of reactivation of mutant p53 in cancer cells. In cancer cells carrying the Y220C mutant, PK7242 binds to the p53-Y220C core domain and induces growth inhibition, cell-cycle arrest, and apoptosis.
|
-

- HY-15003
-
|
|
FLT3
Apoptosis
|
Cancer
|
|
ATH686 is a potent, selective and ATP-competitive FLT3 inhibitor. ATH686 target mutant FLT3 protein kinase activity and inhibit the proliferation of cells harboring FLT3 mutants via induction of apoptosis and cell cycle inhibition. ATH686 has antileukemic effects .
|
-

- HY-78869
-
|
Osimertinib analog
|
EGFR
|
Cancer
|
|
Mutated EGFR-IN-1 (Osimertinib analog) (Intermediate 168) is an intermediate of the mutant EGFR inhibitor.
|
-

- HY-100020
-
-

- HY-18690A
-
-

- HY-162441
-
|
|
Ras
|
Cancer
|
|
pan-KRAS-IN-8 (Compound 38) is an inhibitor for in human tumor mutated genes KRAS, which inhibits proliferation of KRAS mutated cells AsPC-1 (G12D mutant) and SW480 (G12V mutant) with IC50 of 0.07 and 0.18 nM, respectively .
|
-

- HY-162440
-
|
|
Ras
|
Cancer
|
|
pan-KRAS-IN-7 (Compound 25) is an inhibitor for in human tumor mutated genes KRAS, which inhibits proliferation of KRAS mutated cells AsPC-1 (G12D mutant) and SW480 (G12V mutant) with IC50 of 0.35 and 0.51 nM, respectively .
|
-

- HY-162442
-
|
|
Ras
|
Cancer
|
|
pan-KRAS-IN-9 (Compound 52) is an inhibitor for in human tumor mutated genes KRAS, which inhibits proliferation of KRAS mutated cells AsPC-1 (G12D mutant) and SW480 (G12V mutant) with IC50 of 0.24 and 0.30 nM, respectively .
|
-

- HY-153306
-
|
RLY-2608
|
PI3K
|
Cancer
|
|
Zovegalisib (RLY-2608) is an orally active first-in-class allosteric mutant-selective inhibitor of PI3Ka with anti-tumor activity. Zovegalisib inhibits tumor growth in PIK3CA-mutant xenograft mice models with minimal impact on insulin .
|
-

- HY-162443
-
|
|
Ras
|
Cancer
|
|
pan-KRAS-IN-10 (Compound 58) is an inhibitor for in human tumor mutated genes KRAS, which inhibits proliferation of KRAS mutated cells AsPC-1 (G12D mutant) and SW480 (G12V mutant) with IC50 of 0.7 and 0.24 nM, respectively .
|
-

- HY-122578
-
|
|
MDM-2/p53
|
Cancer
|
|
P53R3 is a potent p53 reactivator and restores sequence-specific DNA binding of p53 hot spot mutants, including p53 R175H, p53 R248W and p53 R273H. P53R3 induces p53-dependent antiproliferative effects with much higher specificity than PRIMA-1. P53R3 enhances the recruitment of wild-type p53 and p53 M237I to several target gene promoters. P53R3 strongly enhances the mRNA, total protein and cell surface expression of the death receptor death receptor 5 (DR5). P53R3 is used for cancer research .
|
-

- HY-W746957
-
-

- HY-W753726
-
-

- HY-161030
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-92 (compound 15) is an allosteric T790M/L858R double mutant EGFR inhibitor. EGFR-IN-92 shows antiproliferative activity against H1975 non-small lung cancer (NSCLC) cancer cells expressing double mutant EGFR .
|
-

- HY-19980
-
|
APR-246; PRIMA-1Met
|
MDM-2/p53
Autophagy
Apoptosis
Ferroptosis
|
Cancer
|
|
Eprenetapopt (APR-246) is a first-in-class, small molecule that restores wild-type p53 functions in TP53-mutant cells. Eprenetapopt triggers apoptosis in tumor cells. Eprenetapopt also targets the selenoprotein thioredoxin reductase 1 (TrxR1), a key regulator of cellular redox balance .
|
-

- HY-121161C
-
|
|
Phytohormone
|
Metabolic Disease
|
|
Brassinazole (0.5, 1, 5 μM) causes markedly deformed seedlings, whose morphology is similar to that of BR-deficient mutants. Brassinazole causes cress dwarfism, altering leaf morphology such as the typical downward curl and dark green appearance of Arabidopsis BR-deficient mutants. However, administration of 10 nM BR reversed dwarfism .
|
-

- HY-153008
-
|
|
Bcr-Abl
|
Cancer
|
|
BCR-ABL-IN-7 (compound 4) is a WT and T315I mutant ABL kinases inhibitor. BCR-ABL-IN-7 effectively inhibits activities of WT and T315I mutant ABL kinases. BCR-ABL-IN-7 can be used for the research of chronic myeloid leukemia (CML) research .
|
-

- HY-156671B
-
|
|
Ras
PI3K
ERK
mTOR
Apoptosis
|
Cancer
|
|
RMC-4998 TFA is an orally active inhibitor targeting the active or GTP-bound state of the KRAS G12C mutant. RMC-4998 TFA can form a ternary complex with intracellular CYPA and the activated KRAS G12C mutant, with an IC50 value of 28 nM. RMC-4998 TFA can inhibit ERK signaling in KRAS G12C mutant cancer cells and induce apoptosis. RMC-4998 TFA can be used for for non-small cell lung cancer (NSCLC) research .
|
-

- HY-156671A
-
|
|
Ras
PI3K
ERK
mTOR
Apoptosis
|
Cancer
|
|
RMC-4998 formic is an orally active inhibitor targeting the active or GTP-bound state of the KRAS G12C mutant. RMC-4998 formic can form a ternary complex with intracellular CYPA and the activated KRAS G12C mutant, with an IC50 value of 28 nM. RMC-4998 formic can inhibit ERK signaling in KRAS G12C mutant cancer cells and induce apoptosis. RMC-4998 formic can be used for non-small cell lung cancer (NSCLC) research .
|
-

- HY-175025
-
|
|
PROTACs
Ras
Apoptosis
|
Cancer
|
|
CH091138 is a potent and selective KRASG12D PROTAC degrader with DC50s of 148.3 nM in HeLa cells and 469.8 nM in AsPC-1 cells. CH091138 selectively degrades exogenous and endogenous KRASG12D but not KRAS WT or other KRAS mutants (G12C/G12S/G12V), depending on the VHL-mediated ubiquitin-proteasome system. CH091138 exhibits potent anti-tumor activity and induces cancer cell apoptosis. CH091138 can be used for the studies of pancreatic cancer and colon cancer. (Pink: KRASG12D ligand (HY-175144); Blue: VHL E3 ligase ligand (HY-138678); Black: Linker; VHL E3 ligase ligand + Linker (HY-136006B)) .
|
-

- HY-155742
-
|
|
CFTR
|
Others
|
|
CFTR corrector 12 (compound 17C) is a bithiazole derivative, serving as CFTR corrector. CFTR corrector 12 has the ability to correct some folding defective mutants of the channel responsible for the control of chloride transport across the plasma membrane. CFTR corrector 12 recovers the α-sarcoglycan (α-SG) content in mutant cells .
|
-

- HY-122054
-
|
|
Keap1-Nrf2
|
Cancer
|
|
BPK-29 is a specific ligand that disrupts the atypical orphan nuclear receptor NR0B1-protein (such RBM45 and SNW1) interactions by covalently modifying C274. BPK-29 impairs the anchorage-independent growth of KEAP1-mutant cancer cells .
|
-

- HY-176875
-
|
|
Molecular Glues
|
Cancer
|
|
CoREST/LSD1 degrader-1 (Compound 4) is a molecular glue degrader targeting the KBTBD4 E3 ubiquitin ligase. CoREST/LSD1 degrader-1 is promising for research of KBTBD4-mutant driven cancers (e.g., medulloblastoma) and epigenetic dysregulation disorders .
|
-

- HY-122054A
-
|
|
Keap1-Nrf2
|
Cancer
|
|
BPK-29 hydrochloride is a specific ligand that disrupts the atypical orphan nuclear receptor NR0B1-protein (such RBM45 and SNW1) interactions by covalently modifying C274. BPK-29 hydrochloride impairs the anchorage-independent growth of KEAP1-mutant cancer cells .
|
-

- HY-167690
-
|
|
Endogenous Metabolite
|
Infection
|
|
MK-6186 is a novel non-nucleoside reverse transcriptase inhibitor with sub-nanomolar activity against wild-type viruses and the two most common NNRTI-resistant RT mutants (K103N and Y181C). MK-6186 exhibits excellent antiviral activity against K103N and Y181C mutant viruses. When MK-6186 targets 12 common NNRTI-associated mutant viruses, only two relatively rare mutants (Y188L and V106I/Y188L) show high resistance, with FC values exceeding 100, while the FC values of the remaining viruses are all below 10. In addition, when MK-6186 faces 96 clinical virus isolates carrying NNRTI-resistant mutations, most (70%) viruses show more than 10-fold resistance to efavirenz (EFV), while only 29% of mutant viruses show more than 10-fold resistance to MK-6186 .
|
-

- HY-19896
-
COTI-2
4 Publications Verification
|
MDM-2/p53
Apoptosis
|
Cancer
|
|
COTI-2, an anti-cancer agent with low toxicity, is an orally available third generation activator of p53 mutant forms. COTI-2 acts both by reactivating mutant p53 and inhibiting the PI3K/AKT/mTOR pathway. COTI-2 induces apoptosis in multiple human tumor cell lines. COTI-2 exhibits antitumor activity in HNSCC through p53-dependent and -independent mechanisms. COTI-2 converts mutant p53 to wild-type conformation .
|
-

- HY-164469
-
|
|
Bcr-Abl
Apoptosis
STAT
|
Cancer
|
|
CHMFL-48 is an orally active BCR-ABL kinase inhibitor targeting both wild-type (wt) and various imatinib-resistant mutants. The IC50 values for CHMFL-48 against ABL wild-type and ABL T315I mutant are 1 nM and 0.8 nM, respectively. CHMFL-48 exerts its effects by blocking the autophosphorylation of BCR-ABL wild-type and mutant forms, which impacts downstream signaling mediators such as STAT5 and CRKL, leading to cell cycle arrest and induction of apoptosis. CHMFL-48 holds potential for research into chronic myeloid leukemia (CML) .
|
-

- HY-P5192
-
|
|
Ras
|
Cancer
|
|
KRAS G12D 8-16 is a mutant KRAS G12D 8-16 peptide .
|
-

- HY-N3492
-
|
|
Others
|
Others
|
|
Isoderrone is a new isoflavone isolated from the methanol extractives from white lupin (cv. Kievskij Mutant) roots .
|
-

- HY-13897
-
|
|
EGFR
|
Cancer
|
|
CNX-2006 is a mutant-selective and irreversible EGFR inhibitor with an IC50 below 20 nM for EGFR T790M.
|
-

- HY-N3496
-
|
|
Others
|
Others
|
|
Isochandalone is a new isoflavone isolated from the methanol extractives from white lupin (cv. Kievskij Mutant) roots .
|
-

- HY-139891
-
|
|
Trk Receptor
|
Cancer
|
|
Trk-IN-6 shows excellent in vitro potency on a panel of TRK mutants (IC50 = 0.2-0.7 nM).
|
-

- HY-12408
-
|
|
Ras
|
Cancer
|
|
6H05 is a selective, and allosteric inhibitor of oncogenic mutant K-Ras(G12C).
|
-

- HY-124070
-
|
|
MDM-2/p53
|
Cancer
|
|
ISA27 is a small-molecule MDM2-p53 protein-protein interaction inhibitor. ISA27 inhibits MDM2-mediated p53 ubiquitination and degradation. ISA27 is promising for research of p53-mutant solid tumors (e.g., thyroid, breast cancer) .
|
-

- HY-154313
-
|
Clospirazine
|
Ras
|
Cancer
|
|
Spiclomazine (Clospirazine) is a potent mutant KRAS(G12C) inhibitor that selectively inhibits mutant KRAS-driven pancreatic cancer. Spiclomazine can eliminate KRas-GTP levels in KRAS-driven pancreatic cancer and effectively inhibit RAS-mediated signaling. Spiclomazine significantly inhibits tumor progression in mouse renal capsule xenotransplantation models .
|
-

- HY-16379
-
|
SB1518
|
JAK
FLT3
|
Cancer
|
|
Pacritinib (SB1518) is a potent inhibitor of both wild-type JAK2 (IC50=23 nM) and JAK2 V617F mutant (IC50=19 nM). Pacritinib also inhibits FLT3 (IC50=22 nM) and its mutant FLT3 D835Y (IC50=6 nM).
|
-

- HY-143729
-
-

- HY-161906
-
|
|
Acyltransferase
|
Cancer
|
|
SD-066-4 is an orally active acyltransferase inhibitor. SD-066-4 can be used in cancer research .
|
-

- HY-151571
-
|
|
Ras
|
Cancer
|
|
ZG1077 is a covalent KRAS G12C inhibitor. ZG1077 can be used in the research of non-small cell lung cancer (NSCLC) .
|
-

- HY-159476
-
|
|
Ras
|
Cancer
|
|
KRAS inhibitor-26 (compound 194a) is a potent KRAS G12V inhibitor (IC50: ≤100 nM). KRAS inhibitor-26 can be used for cancer research .
|
-

- HY-12408A
-
|
|
Ras
|
Cancer
|
|
6H05 TFA is a selective, and allosteric inhibitor of oncogenic mutant K-Ras(G12C).
|
-

- HY-119177
-
|
|
Bcr-Abl
|
Others
|
|
PD173956 is a pyridopyrimidine Bcr-Abl kinase inhibitor with high selectivity for the P-loop mutant form of Bcr-Abl .
|
-

- HY-19885
-
|
|
Bacterial
Dihydrofolate reductase (DHFR)
|
Infection
|
|
AR-102 has inhibitory activity towards Staphylococcus aureus. AR-102 exhibits a competitive potent inhibition of the F98Y mutant DHFR (Ki = 0.22 nM). AR-102 has been determined as ternary complex with NADPH in both wild-type S. aureus DHFR and the TMP-resistant F98Y mutant enzyme .
|
-

- HY-156967
-
|
|
MDM-2/p53
|
Cancer
|
|
BAY 1892005 is a regulator of p53 protein and acts on p53 condensates without causing mutant p53 reactivation .
|
-

- HY-134385
-
|
|
CFTR
|
Others
|
|
6-PhEt-dATP is an ATP analog that specifically enhances the function of certain CFTR mutants and is useful for studying cystic fibrosis .
|
-

- HY-169728
-
-

- HY-153342
-
|
ARV-766
|
PROTACs
Androgen Receptor
|
Cancer
|
|
Luxdegalutamide (ARV-766) is an orally active protein hydrolysis targeted chimeric (PROTAC) targeting androgen receptor (AR), which can degrade AR resistance related mutants, including T878/H875/L702 mutants. Luxdegalutamide has anti-tumor activity and can be used in the study of castration resistant prostate cancer .
|
-

- HY-14929
-
|
GR181413A free base; 1-Deoxygalactonojirimycin
|
Glycosidase
|
Others
|
|
Migalastat (GR181413A free base) is an orally active α-galactosidase A molecular chaperone, with an IC50 value of 0.04 μM for human α-Gal A. Migalastat binds to the active site of certain unstable mutant forms of α-galactosidase A, facilitating their transport to the lysosome. After dissociation in the acidic environment, Migalastat enables the mutant α-galactosidase A to exhibit biological activity .
|
-

- HY-14929A
-
|
GR181413A
|
Glycosidase
|
Others
|
|
Migalastat (GR181413A free base) hydrochloride is an orally active α-galactosidase A molecular chaperone, with an IC50 value of 0.04 μM for human α-Gal A. Migalastat binds to the active site of certain unstable mutant forms of α-galactosidase A, facilitating their transport to the lysosome. After dissociation in the acidic environment, Migalastat enables the mutant α-galactosidase A to exhibit biological activity .
|
-

- HY-P11089
-
|
|
MHC
|
Cancer
|
|
TP53 neoepitope is a high-affinity antigenic peptide targeting HLA-A. TP53 neoepitope can triggers CD8 + T cell-mediated killing of TP53-mutant tumor cells. TP53 neoepitope is promising for research of solid tumors harboring TP53 hotspot mutations (e.g., R175H, R273H) .
|
-

- HY-129550
-
|
|
EGFR
|
Cancer
|
|
BI-4020 is a fourth-generation, orally active, and non-covalent EGFR tyrosine kinase inhibitor. BI-4020 inhibits not only the triple mutant EGFR del19 T790M C797S variant (IC50=0.2 nM in BaF3 cell lines) but also the double mutant EGFR del19 T790M and primary mutant EGFR del19 (IC50=1 nM). BI-4020 also shows activity against EGFR wt (IC50=190 nM). BI-4020 shows high kinome selectivity and good DMPK properties .
|
-

- HY-134877
-
|
|
EGFR
ERK
Akt
|
Cancer
|
|
BAY 2476568 is a potent and mutant-selective inhibitor targeting EGFR exon20 insertion variants. BAY 2476568 potently inhibits the kinase activity of EGFR exon20 insertion mutants (insASV, insSVD, insNPG) with IC50 values of 0.09 nM, 0.21 nM, and 0.11 nM, respectively. BAY 2476568 inhibits EGFR (Y1068) phosphorylation and reduces the phosphorylation of ERK1/2 and Akt (S473) in Ba/F3 cells expressing EGFR exon20 insertion mutants (insASV, insSVD). BAY 2476568 can be used for the study of non-small cell lung cancer (NSCLC) driven by EGFR exon20 insertion mutations .
|
-

- HY-18997
-
PLX7904
3 Publications Verification
PB04
|
Raf
|
Cancer
|
|
PLX7904 is a potent and selective BRAF inhibitor, with IC50 of appr 5 nM against BRAF V600E in mutant RAS expressing cells.
|
-

- HY-101522
-
|
|
EGFR
BMX Kinase
Btk
MEK
|
Cancer
|
|
CHMFL-EGFR-202 is a potent, irreversible inhibitor of epidermal growth factor receptor (EGFR) mutant kinase, with IC50s of 5.3 nM and 8.3 nM for drug-resistant mutant EGFR T790M and WT EGFR kinases, respectively. CHMFL-EGFR-202 exhibits ~10-fold selectivity for EGFR L858R/T790M against the EGFR wild-type in cells. CHMFL-EGFR-202 adopts a covalent “DFG-in-C-helix-out” inactive binding conformation with EGFR, with strong antiproliferative effects against EGFR mutant-driven nonsmall-cell lung cancer (NSCLC) cell lines .
|
-

- HY-N16232
-
|
2-α-Carboxy cholestanol
|
Hedgehog
|
Metabolic Disease
|
|
2-ACC (2-α-Carboxy cholestanol) is a cholestanol, is a mutant Sonic Hedgehog protein D46A activator with an EC50 of ~5 μM.
|
-

- HY-160167
-
|
|
Btk
|
Inflammation/Immunology
|
|
Birelentinib (compound cpd15) is an inhibitor of Bruton's tyrosine kinase (BTK) and mutant BTK with IC50 values of 6.72 and 6.11 nM, respectively .
|
-

- HY-W002798
-
-

- HY-10159A
-
|
AMN107 monohydrochloride monohydrate
|
Bcr-Abl
Autophagy
|
Cancer
|
|
Nilotinib monohydrochloride monohydrate is a second generation tyrosine kinase inhibitor (TKI), is significantly potent against BCR-ABL, and is active against many BCR-ABL mutants.
|
-

- HY-148439
-
|
RMC-6236; RAS-IN-2
|
Ras
PERK
|
Cancer
|
|
Daraxonrasib (RMC-6236) is an orally active, non-covalent RAS (ON) inhibitor. Daraxonrasib disrupts the interaction of wild-type or mutant RAS proteins with the RAS binding domain of BRAF, with EC50 values ranging from 28-220 nM for wild-type KRAS, NRAS, HRAS, and multiple oncogenic RAS variants. Daraxonrasib inhibits pERK. Daraxonrasib has anti-tumor activity against KRAS mutant tumors .
|
-

- HY-157389
-
|
|
Bcr-Abl
|
Cancer
|
|
AK-HW-90 (compound 2B) is a potent panBcr-Abl inhibitor with significant inhibitory activity against Imatinib (HY-15463)-resistant mutants. AK-HW-90 inhibits the resistance mutant Bcr-Abl T315I with an IC50 of 0.65 nM. AK-HW-90 has potential anticancer activity and can be used in the study of chronic myelogenous leukemia (CML) .
|
-

- HY-124153
-
|
|
Btk
|
Cardiovascular Disease
Inflammation/Immunology
|
|
GNE-431 is a potent, selective and noncovalent “pan-BTK” inhibitor against C481R, T474I and T474Ms mutants. GNE-431 shows potency against wild-type BTK (IC50= 3.2 nM) and potency against C481S mutant (IC50= 2.5 nM). GNE-431 is proming for rasearch of haematological disorders and autoimmune diseases .
|
-

- HY-146108
-
|
|
RSV
|
Infection
|
|
RSV-IN-5 (Compound 4) is a potent dual inhibitor of wild-type and mutant respiratory syncytial virus (RSV) fusion proteins. RSV-IN-5 exhibits potent anti-RSV activities against not only wild-type A2 F protein (EC50=2.0 nM), but also D486N-mutant F protein (EC50=8.1 nM) .
|
-

- HY-157229
-
STX-721
1 Publications Verification
|
EGFR
ERK
|
Cancer
|
|
STX-721 is an orally active, irreversible, covalent EGFR exon 20 insertion (ex20ins) inhibitor that selectively targets ex20ins-mutant dynamic protein states. STX-721 potently inhibits the kinase activity of EGFR ex20ins mutants (NPG, ASV, SVD). STX-721 inhibits phosphorylation of EGFR (pEGFR Y1068) and downstream ERK (pERK Thr202/Tyr204), and suppresses proliferation of ex20ins-mutant Ba/F3 cells and human NSCLC cell lines (NCI-H2073 ASV KI, CUTO-14 ASV). STX-721 induces tumor regression in EGFR ex20ins-mutant PDX/CDX models. STX-721 can be used for the study of non-small cell lung cancer (NSCLC) harboring EGFR or HER2 ex20ins mutations .
|
-

- HY-112929
-
|
|
Phosphatase
|
Cancer
|
|
DT-061 is an orally bioavailable activator of protein phosphatase 2A (PP2A) and could be applied in the therapy of KRAS-mutant and MYC-driven tumorigenesis .
|
-

- HY-18082
-
-

- HY-122849
-
|
|
c-Kit
|
Cancer
|
|
CHMFL-KIT-031 is a highly selective KIT V559D inhibitor, with an IC50 of 28 nM. CHMFL-KIT-031 potently affects KIT V559D mutant’s phosphorylation at Y703/719/823 sites. CHMFL-KIT-031 inhibits the tumor growth in BaF3-TEL-KIT-V559D cell inoculated mouse model .
|
-

- HY-W998345
-
|
|
PROTACs
PDK-1
Akt
|
Cancer
|
SMART1 is a highly specific and CRBN-dependent PROTAC that can effectively degrade Smurf1. SMART1 can block the PDK1-Akt signaling pathway in KRAS mutant colorectal cancer. SMART1 can inhibit tumor growth in KRAS mutant colorectal cancer (CRC) xenograft models.(Blue: CRBN ligand; Black: linker; Pink: Smurf1 ligand (Smurf1-L)) .
|
-

- HY-104036
-
IDH-305
2 Publications Verification
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
IDH-305 is an orally available, mutant-selective and brain-penetrant IDH1 inhibitor that targets IDH1 (R132) mutation. IDH-305 exhibits greater than 200 fold selectivity for mutant IDH1 isoforms vs. WT (IC50= 27 nM (IDH1 R132H), 28 nM (IDH1 R132C), 6.14 µM (IDH1 WT)) .
|
-

- HY-145785
-
|
|
MDM-2/p53
Apoptosis
|
Cancer
|
|
ADH-6 is a tripyridylamide compound. ADH-6 abrogates self-assembly of the aggregation-nucleating subdomain of mutant p53 DBD. ADH-6 targets and dissociates mutant p53 aggregates in human cancer cells, which restores p53's transcriptional activity, leading to cell cycle arrest and apoptosis. ADH-6 has the potential for the research of cancer diseases[1].
|
-

- HY-19980G
-
|
APR-246; PRIMA-1Met
|
MDM-2/p53
Apoptosis
Ferroptosis
|
Cancer
|
|
Eprenetapopt (GMP) (APR-246(GMP)) is Eprenetapopt (HY-19980) in GMP grade. Eprenetapopt (APR-246) is a first-in-class, small molecule that restores wild-type p53 functions in TP53-mutant cells. Eprenetapopt triggers apoptosis in tumor cells. Eprenetapopt also targets the selenoprotein thioredoxin reductase 1 (TrxR1), a key regulator of cellular redox balance .
|
-

- HY-16379A
-
|
SB1518 hydrochloride
|
JAK
FLT3
|
Cancer
|
|
Pacritinib hydrochloride is a potent inhibitor of both wild-type JAK2 (IC50=23 nM) and JAK2 V617F mutant (IC50=19 nM). Pacritinib hydrochloride also inhibits FLT3 (IC50=22 nM) and its mutant FLT3 D835Y (IC50=6 nM). Pacritinib hydrochloride can be used for the research of acute myeloid leukemia (AML) and myelofibrosis (MF) .
|
-

- HY-16379R
-
|
SB1518 (Standard)
|
Reference Standards
JAK
FLT3
|
Cancer
|
|
Pacritinib (Standard) is the analytical standard of Pacritinib. This product is intended for research and analytical applications. Pacritinib (SB1518) is a potent inhibitor of both wild-type JAK2 (IC50=23 nM) and JAK2V617F mutant (IC50=19 nM). Pacritinib also inhibits FLT3 (IC50=22 nM) and its mutant FLT3D835Y (IC50=6 nM).
|
-

- HY-161885
-
|
|
Epigenetic Reader Domain
|
Cancer
|
|
SMARCA2-IN-6 is a SMARCA2 (also known as BRM) inhibitor, with IC50s less than 5 nM for SMARCA2 and SMARCA4. SMARCA2-IN-6 inhibits KRT80 gene expression in H1299 cells (IC50: 26 nM). SMARCA2-IN-6 inhibits proliferation in BRG1-mutant SKMEL5 cells (IC50: 13 nM) .
|
-

- HY-160020
-
|
|
Androgen Receptor
|
Cancer
|
|
ET516 is a potent inhibitor of Androgen Receptor. ET516 significantly inhibits the proliferation and tumor growth of prostate cancer cells expressing AR-resistant mutants .
|
-

- HY-U00416
-
|
|
Ras
|
Cancer
|
|
ARS-1323 (Example 30, No. 227) is the racemate of ARS-1620. ARS-1323 is a novel inhibitor of mutant K-ras G12C .
|
-

- HY-154959
-
|
|
Ras
|
Cancer
|
|
AZD4747 is a selective, blood-brain barrier-permeable mutant GTPase KRAS G12C inhibitor. AZD4747 has the potential to study cancer .
|
-

- HY-111050
-
|
|
c-Met/HGFR
|
Cancer
|
|
JNJ-38877618 is a potent, highly selective, orally bioavailable Met kinase inhibitor with IC50s of 2 and 3 nM for wild type and mutant Met, respectively.
|
-

- HY-145126
-
|
|
CFTR
|
Inflammation/Immunology
|
|
CP-628006, a small molecule CFTR potentiator, restores ATP-dependent channel gating to the cystic fibrosis mutant G551D-CFTR.
|
-

- HY-157526
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
EGFR-TK-IN-1 (compound 7o) is a potent mutant EGFR inhibitor with IC50 of 8.5 nM an 9.3 nM against EGFR L858R/T790M and EGFR Del19.EGFR-TK-IN-1 showes strong antiproliferative effects against EGFR mutant-driven non-small cell lung cancer (NSCLC) cells and induces cell apoptosis .
|
-

- HY-145785A
-
|
|
MDM-2/p53
Apoptosis
|
Cancer
|
|
ADH-6 TFA is a tripyridylamide compound. ADH-6 abrogates self-assembly of the aggregation-nucleating subdomain of mutant p53 DBD. ADH-6 TFA targets and dissociates mutant p53 aggregates in human cancer cells, which restores p53's transcriptional activity, leading to cell cycle arrest and apoptosis. ADH-6 TFA has the potential for the research of cancer diseases .
|
-

- HY-161802
-
|
|
Bacterial
|
Infection
|
|
Antibacterial agent 228 (Compound 8) inhibits the mycobacterial ribosome (IC50 for Mycobacterium smegmatis is 2.31 μM) and exhibits antibacterial activity against M. tuberculosis H37Rv (MIC=2 and 0.25 μg/mL for wildtype and Δ1258c mutant), M. abscessus ATCC 19977 (MIC=8 and 8 μg/mL for wildtype and Δ2780c mutant) and M. smegmatis (MIC=8 μg/mL) .
|
-

- HY-W494890
-
|
|
Epigenetic Reader Domain
|
Cancer
|
|
SMARCA2-IN-10 (Compound 4) is a highly selective SMARCA2 ATPase domain inhibitor (IC50=17.676 μM). SMARCA2-IN-10 induces cell death in SMARCA4-deficient tumors. SMARCA2-IN-10 is promising for research of SMARCA4-mutant non-small cell lung cancer, small cell ovarian carcinoma, and melanoma .
|
-

- HY-161819
-
|
|
Trk Receptor
|
Cancer
|
|
TRK-IN-29 (Compound B31) is a second-generation TRK inhibitor (IC50: 9 nM, 0.6 nM, 18 nM, 5 nM, 6 nM for TRKA G595R, TRKA F589L, TRKA G667C, TRKA and TRKC respectively). TRK-IN-29 inhibits the phosphorylation of TRKA. TRK-IN-29 has good antiproliferative activities against NTRK fusion positive cells. TRK-IN-29 has exellent plasma stability and moderate pharmacokinetic properties .
|
-

- HY-P0222
-
|
|
PKA
|
Others
|
|
PKI(5-24) is a potent, competitive, and synthetic peptide inhibitor of PKA (cAMP-dependent protein kinase), with a Ki of 2.3 nM. PKI(5-24) corresponds to residues 5-24 in the naturally occurring heat-stable protein kinase inhibitor .
|
-

- HY-P0222A
-
|
|
PKA
|
Others
|
|
PKI(5-24) TFA is a potent, competitive, and synthetic peptide inhibitor of PKA (cAMP-dependent protein kinase), with a Ki of 2.3 nM. PKI(5-24) TFA corresponds to residues 5-24 in the naturally occurring heat-stable protein kinase inhibitor .
|
-

- HY-P3277
-
|
|
Ras
|
Cancer
|
|
KRpep-2d is a potent K-Ras inhibitor and inhibits proliferation of K-Ras-driven cancer cells. KRpep-2d can be used for cancer research .
|
-

- HY-134904
-
|
RM-006
|
mTOR
|
Cancer
|
|
RMC-6272 (RM-006) is a bi-steric mTORC1-selective inhibitor. RMC-6272 exhibits potent and selective (> 10-fold) inhibition of mTORC1 over mTORC2. RMC-6272 shows improved inhibition of mTORC1 in comparison to Rapamycin, and induces more cell death in TSC2 null tumors .
|
-

- HY-161656
-
|
|
PROTAC Linkers
|
Cancer
|
|
PIP-C-3-Azaspiro[5.5]undecane-boc is the linker for PROTAC. PIP-C-3-Azaspiro[5.5]undecane-boc is utilized for synthesis of PROTAC SOS1 degrader-10 (HY-161654) .
|
-
![PIP-C-3-Azaspiro[5.5]undecane-boc](//file.medchemexpress.eu/product_pic/hy-161656.gif)
- HY-161655
-
-

- HY-P3277A
-
|
|
Ras
|
Cancer
|
|
KRpep-2d (TFA) is a potent K-Ras inhibitor and inhibits proliferation of K-Ras-driven cancer cells. KRpep-2d can be used for cancer research .
|
-

- HY-P4391
-
-

- HY-107977
-
-

- HY-148901
-
|
|
SOD
|
Neurological Disease
|
|
CMB-087229 is a mutant superoxide dismutase 1 (SOD1) protein aggregation inhibitor with IC50 of 67 nM, which can be used in the research of amyotrophic lateral sclerosis .
|
-

- HY-N3920
-
|
|
Bacterial
|
Infection
|
|
Gancaonin G is a 6-prenylated isoflavanone that can be isolated from Glycyrrhiza uralensis. Gancaonin G has antibacterial activity against Streptococcus mutants and MRSA strains .
|
-

- HY-128589
-
|
|
c-Kit
|
Cancer
|
|
CHMFL-KIT-033 is a potent and selective inhibitor of c-KIT T670I mutant for gastrointestinal stromal tumors (GISTs), with an IC50 of 0.045 μM .
|
-

- HY-12090
-
|
MK-0859
|
PCSK9
|
Cardiovascular Disease
|
|
Anacetrapib is a potent CETP inhibitor, with IC50s of 7.9±2.5 nM and 11.8±1.9 nM for rhCETP and C13S CETP mutant, respectively.
|
-

- HY-112802
-
|
|
c-Kit
|
Cancer
|
|
AZD3229 is a potent pan-KIT mutant inhibitor for the treatment of gastrointestinal stromal tumors. AZD3229 inhibits c-KIT with an IC50 value of 223.3 nM .
|
-

- HY-18690AR
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Enasidenib (mesylate) (Standard) is the analytical standard of Enasidenib (mesylate). This product is intended for research and analytical applications. Enasidenib mesylate is a first-in-class, oral, potent, reversible, selective inhibitor of the IDH2 mutant enzymes.
|
-

- HY-153361
-
|
|
PROTACs
Epigenetic Reader Domain
|
Cancer
|
|
YD23 is a selective SMARCA2 PROTAC degrader with DC50 values of 64 nM and 297 nM in H1792 cells and H1975 cells. YD23 induces degradation of SMARCA2, which is synthetic lethal to SMARCA4. YD23 reduces chromatin accessibility only in SMARCA4 deficient cells, including cell cycle and cell growth regulatory genes. YD23 selectively inhibits growth of SMARCA4 mutant lung cancer cells. YD23 has potent tumor growth inhibitory activity in SMARCA4-mutant xenografts. YD23 can be used for the study of non-small cell lung cancer (NSCLC) (Pink: SMARCA2 ligand (HY-44012); Blue: CRBN ligand (HY-41547); Black: Linker (HY-175566)) .
|
-

- HY-120885
-
|
|
Ras
|
Cancer
|
|
(+)-Oxanthromicin (Compound 1) mislocalizes the oncogenic mutant K-Ras from the plasma membrane of intact Madin-Darby canine kidney (MDCK) cells, and exhibits thereby antitumor efficacy .
|
-

- HY-100002
-
-

- HY-18343A
-
|
|
MDM-2/p53
|
Cancer
|
|
CP-31398 dihydrochloride stabilizes the active conformation of p53 and promotes p53 activity in cancer cell lines with mutant or wild-type p53 .
|
-

- HY-18767A
-
|
(R,S)-AG-120
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
(R,S)-Ivosidenib ((R,S)-AG-120) is a less active enantiomer of ivosidenib (AG-120). Ivosidenib can be used in the research of IDH1 mutant cancers .
|
-

- HY-U00417
-
|
|
Ras
|
Cancer
|
|
ARS-1630, a less active enantiomer of ARS-1620, is a novel inhibitor of mutant K-ras G12C extracted from patent WO 2015054572 A1.
|
-

- HY-114778
-
|
SHR3162; Fuzuloparib
|
PARP
|
Cancer
|
|
Fluzoparib (SHR3162) is a potent and orally active PARP1 inhibitor (IC50=1.46±0.72 nM, a cell-free enzymatic assay) with superior antitumor activity. Fluzoparib selectively inhibits the proliferation of homologous recombination repair (HR)-deficient cells, and sensitizes both HR-deficient and HR-proficient cells to cytotoxic agents. Fluzoparib exhibits good pharmacokinetic properties in vivo and can be used for BRCA1/2-mutant relapsed ovarian cancer research .
|
-

- HY-16379S
-
|
SB1518-d4
|
Isotope-Labeled Compounds
JAK
FLT3
|
Cancer
|
|
Pacritinib-d8 (SB1518-d8) is the deuterium labeled Pacritinib (HY-16379). Pacritinib (SB1518) is a potent inhibitor of both wild-type JAK2 (IC50=23 nM) and JAK2 V617F mutant (IC50=19 nM). Pacritinib also inhibits FLT3 (IC50=22 nM) and its mutant FLT3 D835Y (IC50=6 nM).
|
-

- HY-117540
-
|
|
Histone Methyltransferase
|
Cancer
|
|
ZLD10A is a highly potent and selective EZH2 inhibitor with the activity of inhibiting H3K27 methylation. ZLD10A can inhibit wild-type and mutant EZH2 with nanomolar potency and has more than 1000-fold selectivity for the other 10 histone methyltransferases. ZLD10A inhibited cell proliferation of DLBCL cell lines in a concentration- and time-dependent manner, showing a potential antiproliferative effect. ZLD10A can be used in the study of EZH2 mutant lymphomas .
|
-

- HY-148404
-
|
|
MDM-2/p53
|
Cancer
|
|
p53 Activator 5 (compound 134A) is a potent p53 activator with a SC150 value of <0.05 mM. p53 Activator 5 can bind to mutant p53 and restore the ability of the p53 mutant to bind DNA. p53 Activator 5 shows anti-tumor activity . p53 Activator 5 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-136379
-
|
Cdc42-IN-1
|
Ras
|
Cancer
|
|
CID44216842 (Cdc42-IN-1) is a potent Cdc42-selective guanine nucleotide binding lead inhibitor. The EC50s for Cdc42 WT and Cdc42Q61L mutant are 1.0 and 1.2 μM in GTP binding assay, respectively. The EC50s for Cdc42 WT and Cdc42Q61L mutant are 0.3 and 0.5 μM in GDP binding assay, respectively. Use as a molecular probe .
|
-

- HY-148402
-
|
|
MDM-2/p53
|
Cancer
|
|
p53 Activator 3 (compound 87A) is a potent p53 activator with a SC150 value of <0.05 mM. p53 Activator 3 can bind to mutant p53 and restore the ability of the p53 mutant to bind DNA. p53 Activator 3 shows anti-tumor activity . p53 Activator 3 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-16379B
-
|
SB1518 citrate
|
JAK
FLT3
|
Cancer
|
|
Pacritinib (SB1518) citrate is a potent inhibitor of both wild-type JAK2 (IC50=23 nM) and JAK2 V617F mutant (IC50=19 nM). Pacritinib citrate also inhibits FLT3 (IC50=22 nM) and its mutant FLT3 D835Y (IC50=6 nM). Pacritinib citrate can be used for the research of acute myeloid leukemia (AML) and myelofibrosis (MF) .
|
-

- HY-150699
-
|
|
Bacterial
|
Infection
|
|
TXA6101 is a bacterial protein FtsZ (filamentous temperature-sensitive protein Z) inhibitor that inhibits bacterial division. TXA6101 has antimicrobial activity against MRSA isolates expressing either the G193D or G196S mutant FtsZ with the MIC value of 1 μg/mL, retains significant activity against the TXA707-resistant FtsZ mutant. TXA6101 can be used as a potential method against Gram-negative bacterial infections .
|
-

- HY-13810
-
|
|
Raf
|
Cancer
|
|
PF-04880594 is a potent and selective RAF inhibitor. PF-04880594 inhibits both wild-type and mutant BRAF and CRAF. PF-04880594 shows antitumor activity .
|
-

- HY-18205
-
-

- HY-168876
-
|
|
EGFR
|
Cancer
|
|
AZ14240475 is a potent, selective, and brain-penetrant inhibitor of EGFR Ex20Ins mutants, with the pIC50 of 7.6. AZ14240475 plays an important role in cancer research .
|
-

- HY-N5141
-
|
|
Others
|
Others
|
|
Isoscoparin-2′′O-glucoside is a flavonoid that can be found in yellow grain mutant of rice. Isoscoparin-2′′O-glucoside shows antioxidant activity .
|
-

- HY-121262
-
|
Bisabolene; γ-Bisabolene
|
Others
|
Others
|
|
Bisabolene is a compound that can be synthesized by engineered bacteria. Bisabolene can be synthesized by heterologous expression of related genes in cyanobacteria. Its production and changes in related metabolites were studied in glycogen-deficient mutants.
|
-

- HY-108637
-
|
|
MDM-2/p53
|
Cancer
|
|
PhiKan 083 is a carbazole derivative, which binds to the surface cavity and stabilizes Y220C (a p53 mutant), with a Kd of 167 μM. PhiKan 083 can be used for cancer research .
|
-

- HY-176041
-
|
|
Calcium Channel
|
Cardiovascular Disease
|
|
Troponin modulator 1 (compound 7) modulates the mutant cardiac muscle thin filament function. Troponin modulator 1 enhances Ca 2+ sensitivity by phosphorylation with an EC50 of 67 μM .
|
-

- HY-108018
-
|
|
HCV Protease
|
Infection
|
|
MK 6325 is a HCV NS3/4a protease inhibitor. MK 6325 displays the broad genotype and mutant potency. MK 6325 can be used for the research of infection .
|
-

- HY-111079
-
|
|
HIV
Reverse Transcriptase
|
Others
|
|
HIV-1 inhibitor-70 (compound 20) is a bifunctional inhibitor that inhibits both wild-type (WT) and K103N mutant reverse transcriptases (RTs) of HIV-1.
|
-

- HY-135805A
-
|
|
EGFR
|
Cancer
|
|
(Rac)-JBJ-04-125-02 is the racemate of JBJ-04-125-02. JBJ-04-125-02 is a potent, mutant-selective, allosteric and orally active EGFR inhibitor .
|
-

- HY-149711
-
|
|
ICMT
|
Metabolic Disease
|
|
Farnesylcysteine (FC) is a competitive inhibitor of ICMT. The fcly mutant has quantitatively low farnesylcysteine (FC) lyase activity and an enhanced response to ABA. Farnesylcysteine induces an ABA hypersensitive phenotype in Arabidopsis thaliana .
|
-

- HY-13237
-
-

- HY-18604
-
|
|
Ras
|
Cancer
|
|
K-Ras G12C-IN-1 is a novel and irreversible inhibitor of mutant K-ras G12C extracted from patent WO 2014152588 A1.
|
-

- HY-N14782
-
|
|
Antibiotic
Bacterial
|
Infection
Cancer
|
|
10-Decarbomethoxyaclacinomycin A is an anthracycline antibiotic can be produced by Streptomyces galilaeus MA144-Mlt mutant strain KE303. 10-Decarbomethoxyaclacinomycin A has antibacterial activities .
|
-

- HY-153007
-
|
|
Dihydrofolate reductase (DHFR)
|
Infection
|
|
DHFR-IN-5 is a potent and orally active dihydrofolate reductase (DHFR) inhibitor with a Ki value of 0.54 nM for quadruple mutant Plasmodium falciparum DHFR. DHFR-IN-5 shows anti-malaria activity .
|
-

- HY-153007A
-
|
|
Dihydrofolate reductase (DHFR)
|
Infection
|
|
DHFR-IN-5 hydrochloride is a potent and orally active dihydrofolate reductase (DHFR) inhibitor with a Ki value of 0.54 nM for quadruple mutant Plasmodium falciparum DHFR. DHFR-IN-5 hydrochloride shows anti-malaria activity .
|
-

- HY-W740650
-
|
GR181413A-15N
|
Isotope-Labeled Compounds
Glycosidase
|
Others
|
|
Migalastat hydrochloride- 15N (GR181413A- 15N) is the 15N-labeled Migalastat hydrochloride (HY-14929A). Migalastat (GR181413A free base) hydrochloride is an orally active α-galactosidase A molecular chaperone, with an IC50 value of 0.04 μM for human α-Gal A. Migalastat binds to the active site of certain unstable mutant forms of α-galactosidase A, facilitating their transport to the lysosome. After dissociation in the acidic environment, Migalastat enables the mutant α-galactosidase A to exhibit biological activity .
|
-

- HY-W728096
-
|
GR181413A-d5 free base
|
Isotope-Labeled Compounds
Glycosidase
|
Others
|
|
Migalastat-d5 (GR181413A-d5 (free base)) is deuterium labeled Migalastat. Migalastat (GR181413A free base) is an orally active α-galactosidase A molecular chaperone, with an IC50 value of 0.04 μM for human α-Gal A. Migalastat binds to the active site of certain unstable mutant forms of α-galactosidase A, facilitating their transport to the lysosome. After dissociation in the acidic environment, Migalastat enables the mutant α-galactosidase A to exhibit biological activity .
|
-

- HY-161777
-
|
|
SARS-CoV
Virus Protease
|
Infection
|
|
SARS-CoV-2 Mpro-IN-23 (Compound 2) is an inhibitor for SARS-CoV-2 main protease (Mpro), which inhibits wildtype Mpro and mutant Mpro variants, with IC50 of 0.057-0.92 μM. SARS-CoV-2 Mpro-IN-23 inhibits the post-entry viral processes of wild-type SARS-CoV-2 single-round infectious particles (SRIPs), suppresses the viral replication of Mpro wildtype and Mpro mutants with EC50 of 0.02-0.52 μM .
|
-

- HY-103369
-
|
|
CFTR
|
Endocrinology
|
|
PG01 is a potent CFTR Cl - channel potentiator. PG01 can correct gating defects of CFTR mutants, is effective on b>E193K, G970R and G551D (CFTR mutants) with Kd values of 0.22 μM, 0.45 μM and 1.94 μM, respectively. PG01 is also effective on ΔF508 (Ka of 0.3 μM). PG01 increases ΔF508-CFTR Cl - current after adding Forskolin .
|
-

- HY-P99849
-
|
ABT-806
|
EGFR
|
Cancer
|
|
Depatuxizumab is a brain-penetrant and humanized tumor-specific anti EGFR monoclonal antibody. Depatuxizumab inhibits the growth of xenograft models of mutant EGFRvIII and wild-type EGFR. Depatuxizumab can be used for research on cancer .
|
-

- HY-112256
-
ACP-105
5 Publications Verification
|
Androgen Receptor
|
Inflammation/Immunology
|
|
ACP-105 is an orally available, selective amd potent androgen receptor modulator (SARM), with pEC50s of 9.0 and 9.3 for AR wild type and T877A mutant, respectively.
|
-

- HY-162300
-
|
|
EGFR
|
Cancer
|
EGFR kinase inhibitor 4 (Compound 4) is a bivalent ATP-allosteric EGFR inhibitor (IC50: 1.8 nM for mutant EGFR (LRTMCS)). EGFR kinase inhibitor 4 can be used for research of NSCLC .
|
-

- HY-164315
-
|
|
Ras
|
Cancer
|
|
KRAS G12C inhibitor 67 (example 35) is a KRAS G12C inhibitor that selectively inhibits the growth of many KRAS G12C mutant tumor cell lines .
|
-

- HY-130253
-
|
|
IRAK
|
Cancer
|
|
IRAK4-IN-6 is an orally efficacious and selective IRAK4 inhibitor with an IC50 of 4 nM, and targetes MyD88 L265P mutant diffuse large B cell lymphoma .
|
-

- HY-120122
-
|
|
MDM-2/p53
|
Cancer
|
|
PK7088 is a pyrazole and a specific peptide. PK7088 supports the reactivation of mutant p53 by converting it to a form exhibiting wild-type properties. PK7088 exhibit anticancer activity in cancer research .
|
-

- HY-111627
-
|
|
Nucleoside Antimetabolite/Analog
|
Cancer
|
|
5-Aza-7-deazaguanine is a substrate for wild-type (WT) E. coli purine nucleoside phosphorylase and its Ser90Ala mutant in the synthesis of base-modified nucleosides .
|
-

- HY-163636
-
|
A-442b
|
Phosphodiesterase (PDE)
Ras
|
Cancer
|
|
Deltaflexin3 is a potent PDE6D inhibitor. Deltaflexin3 reduces the signaling of Ras and selectively decreases the KRAS mutant and PDE6D-dependent cancer cells growth .
|
-

- HY-108637A
-
|
|
MDM-2/p53
|
Cancer
|
|
PhiKan 083 hydrochloride is a carbazole derivative, which binds to the surface cavity and stabilizes Y220C (a p53 mutant), with a Kd of 167 μM , and a relative binding affinity (Kd) of 150 μM in Ln229 cells .
|
-

- HY-107415
-
|
|
Raf
|
Cancer
|
|
PLX7922, a RAF inhibitor, can bind with BRAF V600E. PLX7922 inhibits pERK in BRAF V600E cell lines, and activates pERK in mutant NRAS cell lines .
|
-

- HY-18690
-
|
AG-221
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Enasidenib is an oral, potent, reversible, selective inhibitor of the IDH2 mutant enzymes, with IC50s of 100 and 400 nM against IDH2 R140Q and IDH2 R172K, respectively.
|
-

- HY-P2319
-
|
|
p38 MAPK
JNK
|
Inflammation/Immunology
|
|
OVA-E1 peptide, is an antagonist variant of SIINFEKL [OVA (257-264). OVA-E1 peptide, activates the p38 and JNK cascades similarly in mutant and wild-type thymocytes .
|
-

- HY-135914
-
|
|
EGFR
|
Cancer
|
|
JBJ-02-112-05 is a potent, mutant-selective, allosteric and orally active EGFR inhibitor with an IC50 of 15 nM for EGFR L858R/T790M .
|
-

- HY-112306
-
|
DCC-2618
|
c-Kit
PDGFR
FLT3
VEGFR
Apoptosis
|
Cancer
|
|
Ripretinib (DCC-2618) is an orally bioavailable, selective KIT and PDGFRA switch-control inhibitor. Ripretinib (DCC-2618) targets and binds to both wild-type and mutant forms of KIT and PDGFRA specifically at their switch pocket binding sites, thereby preventing the switch from inactive to active conformations of these kinases and inactivating their wild-type and mutant forms. Ripretinib (DCC-2618) also inhibits multiple other kinase targets, such as FLT3 and KDR (or VEGFR-2) . DCC-2618 exerts antineoplastic effect and induces apoptosis .
|
-

- HY-160142
-
|
|
PROTACs
Btk
Syk
MEK
ERK
|
Cancer
|
|
UBX-382 is an orally active BTK PROTAC degrader with a DC50 of 4.56 nM. UBX-382 inhibits B-cell receptor signaling by targeting BTK. UBX-382 shows superior degradation activity for wild-type and mutant BTK proteins. UBX-382 inhibits tumor growth in murine xenograft models harboring wild-type or C481S mutant BTK-expressing TMD-8 cells. UBX-382 can be used for the study of B-cell-related blood cancers .
|
-

- HY-162809
-
|
|
Ras
|
Cancer
|
|
XMU-MP-9 is a bifunctional compound that binds to the C2 domain of Nedd4-1 and the allosteric site of K-Ras. XMU-MP-9 enhances the interaction between Nedd4-1 and K-Ras, induces conformational changes in the Nedd4-1/K-Ras complex, promotes the ubiquitination and degradation of multiple K-Ras mutants, and inhibits the proliferation of cells carrying K-Ras mutants. XMU-MP-9 can be used for the study of colon, lung and pancreatic cancer .
|
-

- HY-112654A
-
|
|
Endogenous Metabolite
|
Cancer
|
|
GCN2iB acetate is a GCN2 inhibitor that enhances Gcn2 activity and Atf4 expression. GCN2iB can activate Gcn2 mutants lacking functional regulatory regions or certain kinase domain substitution mutants. GCN2iB increases eIF2 phosphorylation by Gcn2 at low concentrations, thereby enhancing the cellular response to nutritional stress. GCN2iB may have potential benefits for the inhibition of cancer cells expressing low basal levels of aspartate synthetase, enhancing their sensitivity to the anti-leukemic compound L-aspartase .
|
-

- HY-175351
-
|
|
HIV
Reverse Transcriptase
|
Infection
|
|
HIV-1-IN-83 (Compound 18E) is a HIV-1 non-nucleoside reverse transcriptase inhibitor with an IC50 of 0.45 μM for wild-type HIV-1 non-nucleoside reverse transcriptase. HIV-1-IN-83 has potent antiviral activity against both HIV-1 wild-type and mutant strains and improves antidrug resistance for Y188L and RES056 mutant strains. HIV-1-IN-83 has no significant toxicity up to 11.088 μM .
|
-

- HY-15217
-
|
|
Haspin Kinase
|
Cancer
|
|
CHR-6494 is a potent inhibitor of haspin, with an IC50 of 2 nM. CHR-6494 inhibits histone H3T3 phosphorylation. CHR-6494 can be used in the research of cancer .
|
-

- HY-110350
-
|
|
Haspin Kinase
|
Cancer
|
|
CHR-6494 TFA is a potent inhibitor of haspin, with an IC50 of 2 nM. CHR-6494 TFA inhibits histone H3T3 phosphorylation. CHR-6494 TFA induces the apoptosis of cancer cells, including melanoma and breast cancer. CHR-6494 TFA can be used in the research of cancer .
|
-

- HY-137191
-
|
|
EGFR
|
Cancer
|
|
CH7233163 is a noncovalent ATP-competitive inhibitor for EGFR-Del19/T790M/C797S. CH7233163 can overcome Osimertinib (HY-15772)-Resistant EGFR-Del19/T790M/C797S mutation. CH7233163 blocks the EGFR phosphorylation in the Del19/T790M/C797S_NIH3T3 cells. CH7233163 has antitumor activities .
|
-

- HY-168221
-
|
|
PROTACs
|
Cancer
|
|
YD54 is a PROTAC based SMARCA2 degrader with a DC50 of 3.5 nM (Red: SMARCA2 degrader (HY-44012B), black: linker, Blue: E3 ligase ligand) .
|
-

- HY-132193
-
|
|
RET
|
Cancer
|
|
RET-IN-4 is a potent, selective and orally active RET inhibitor with IC50s of 1.29 nM, 1.97 nM, and 0.99 nM for RET (WT), RET (V804M), and RET (M918T), respectively. RET-IN-4 exhibits better kinases selectivity against JAK2 (IC50 of 4.4 nM) and FLT3 (IC50 of 30.8 nM). RET-IN-4 has anticancer effects .
|
-

- HY-164526
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
SH1573 is an orally active mIDH2 inhibitor. SH1573 has a strong and selective inhibitory effect on mIDH2 R140Q protein (IC50=4.78 nmol/L), and can effectively reduce the production of the carcinogenic metabolite 2-hydroxyglutarate (2-HG) in animal models, cell lines, serum and tumors. SH1573 can be used for the study of acute myeloid leukemia (AML) .
|
-

- HY-142680
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-23 is a potent EGFR TKI (tyrosine kinase inhibitor) with an IC50 of 8.05 nM for BaF3/EGFR-DEL19/T790M/C797S cell (WO2021244502A1, compound 8) .
|
-

- HY-146710
-
|
|
RET
|
Cancer
|
|
RET-IN-16 is a potent and selective RET inhibitor with IC50s of 3.98 nM, 8.42 nM, 15.05 nM, 7.86 nM, 5.43 nM and 8.86 nM for RET(WT), RET(M918T), RET(V804L), RET(V804M), RET-CCDC6 and RET-KIF5B, respectively. RET-IN-16 has anticancer effects .
|
-

- HY-116528
-
|
|
HIV
|
Infection
|
|
GCA-186 is a potent anti-HIV-1 agent. GCA-186 is highly active against both wild type and mutated HIV-1 strains with EC50s of 1, 180, 1, and 40 nM for IIIB, IIIB-R(Y181C), NL4-3 and NL4-3K103N of HIV-1 strains, respectively .
|
-

- HY-161753
-
|
|
CFTR
|
Inflammation/Immunology
|
|
CFTR potentiator 1 (I1421) is a potent CFTR potentiator with an EC50 value of 64 nM. CFTR potentiator 1 allosterically activates a wide range of CF-causing mutants, such as ΔF508 and G551D CFTR .
|
-

- HY-153306A
-
|
(S)-RLY-2608
|
PI3K
Drug Isomer
|
Cancer
|
|
(S)-Zovegalisib ((S)-RLY-2608) is an S-enantiomer of of Zovegalisib (HY-153306). Zovegalisib is an orally active first-in-class allosteric mutant-selective inhibitor of PI3Ka with anti-tumor activity .
|
-

- HY-169294
-
|
|
Sodium Channel
|
Metabolic Disease
|
|
SLC6A8 corrector 1 is an orally active and brain-penetrant mutant SLC6A8 variant corrector. SLC6A8 corrector 1 can be used for the study of creatine transporter deficiency (CTD) .
|
-

- HY-W097867
-
|
8-Methoxy Ciprofloxacin; 8-Methoxy-cipro
|
Bacterial
|
Infection
|
|
3-Desmethyl Gatifloxacin (8-Methoxy Ciprofloxacin) induces DNA cleavage mediated by wild-type gyrase and Quinolone-resistant mutants. 3-Desmethyl Gatifloxacin can be used for research of drug resistance .
|
-

- HY-P2319A
-
|
|
p38 MAPK
JNK
|
Inflammation/Immunology
|
|
OVA-E1 peptide TFA, is an antagonist variant of SIINFEKL [OVA (257-264). OVA-E1 peptide, activates the p38 and JNK cascades similarly in mutant and wild-type thymocytes .
|
-

- HY-P1974
-
|
WF11899A
|
Antibiotic
|
Infection
|
|
FR901379 (WF11899A) is a novel echinocandin-type lipopeptide antibiotic. FR901379 is primarily produced by mutant strain M-7 of Coleophoma empetri F-11899 (FERM BP-2635) .
|
-

- HY-18634
-
|
ZMC1
|
MDM-2/p53
|
Cancer
|
|
NSC319726 (ZMC1) is a mutant p53R175 reactivator; inhibits growth of fibroblasts expressing the p53R175 mutation (IC50 = 8 nM); shows no inhibition for p53 wild-type cells.
|
-

- HY-146243
-
|
|
Ras
Apoptosis
|
Cancer
|
|
TH-Z835 is a mutant selective KRAS (G12D) inhibitor with an IC50 of 1.6 μM. TH-Z835 inhibits both mantGMPPNP/GPPNP exchange and GPPNP/mantGMPPNP exchange .
|
-

- HY-13223A
-
|
CP 868596-26
|
Autophagy
PDGFR
FLT3
|
Cancer
|
|
Crenolanib benzenesulfonate is a potent and selective inhibitor of wild-type and mutant isoforms of the class III receptor tyrosine kinases FLT3 and PDGFRα/β with Kds of 0.74 nM and 2.1 nM/3.2 nM, respectively.
|
-

- HY-B0282
-
|
ACh chloride
|
nAChR
Calcium Channel
Endogenous Metabolite
|
Neurological Disease
Cancer
|
|
Acetylcholine chloride (ACh chloride), a neurotransmitter, is a potent cholinergic agonist. Acetylcholine chloride is a modulator of the activity of dopaminergic (DAergic) neurons through the stimulation of nicotinic acetylcholine receptors (nAChRs) . Acetylcholine chloride inhibits p53 mutant peptide aggregation in vitro .
|
-

- HY-169427
-
|
|
EGFR
|
Cancer
|
|
FRF-06-057 is an ATP-allosteric EGFR inhibitor against wild-type and mutant EGFR, with IC50s of 17 nM (LR/TM), 29 nM (LR/TM/CS), 220 nM (LR), > 1000 nM (WT) respectively .
|
-

- HY-W591307
-
|
|
PROTAC Linkers
|
Cancer
|
|
P131 is a PROTAC molecule comprised of Pomalidomide-based cereblon ligand and IBT6A, linked by a PEG 3 chain. P131 can degrade both wild-type and C481S mutant BTK with nanomolar potencies.
|
-

- HY-13223
-
|
CP-868596
|
FLT3
PDGFR
Autophagy
|
Cancer
|
|
Crenolanib is a potent and selective inhibitor of wild-type and mutant isoforms of the class III receptor tyrosine kinases FLT3 and PDGFRα/β with Kds of 0.74 nM and 2.1 nM/3.2 nM, respectively.
|
-

- HY-111760
-
-

- HY-109061
-
|
YH25448; GNS-1480
|
EGFR
|
Cancer
|
|
Lazertinib (YH25448) is a potent, highly mutant-selective, blood-brain barrier permeable, orally available and irreversible third-generation EGFR tyrosine kinase inhibitor, and can be used in the research of non-small cell lung cancer .
|
-

- HY-173629
-
|
|
Ras
Apoptosis
|
Cancer
|
|
RMC-5127 is an orally active and brain-penetrant mutant-selective tri-complex RAS G12V inhibitor. RMC-5127 non-covalently binds to cyclophilin A (CypA), forming a binary complex that engages RASG12V(ON) to form a high-affinity tri-complex, sterically inhibiting RAS binding . RMC-5127 inhibits the RAS pathway in KRAS G12V mutant cancer cells, reducing cell proliferation and inducing apoptosis. RMC-5127 is promising for research of cancers with RAS mutations, such as non-small cell lung cancer .
|
-

- HY-172964
-
|
|
PI3K
Akt
mTOR
Apoptosis
Reactive Oxygen Species (ROS)
|
Cancer
|
|
KIM-161 is a PIK3CA inhibitor. KIM-161 has significant antiproliferative activity with IC50 values of 1.428 and 1.562 µM against PI3KCA mutant breast cancer MCF7 and T47D cells, respectively. KIM-161 induces apoptosis and cell cycle arrest by inhibiting the PI3K/AKT/mTOR signaling pathway and inducing ROS production. KIM-161 can be used to study breast cancer and its PI3KCA mutant subtypes .
|
-

- HY-153346
-
|
RMC-6291
|
Ras
ERK
Apoptosis
|
Cancer
|
|
Elironrasib is an orally active and covalent inhibitor of KRAS G12C(ON). Elironrasib forms a tri-complex within tumor cells between KRAS G12C(ON) and cyclophilin A (CypA). Thus, Elironrasib prevents KRAS G12C(ON) from signaling via steric blockade of RAS effector binding. Elironrasib inhibits ERK signaling and induced apoptosis in KRASG12C-mutant H358 cells. Elironrasib also inhibits the proliferation of KRAS G12C mutant cells with a median IC50 of 0.11 nM .
|
-

- HY-174136
-
|
|
STING
|
Inflammation/Immunology
|
|
STING Degrader-2 (Compound SI-43) is a STING inhibitor and mutant-specific degrader. STING Degrader-2 can effectively inhibit cGAMP-induced STING activation and significantly reduce the release of IFN-β and CXCL-10. It inhibits the activity of STING by binding to two pockets of the STING dimer and induces proteasome-independent degradation of mutants STING S154 and STING M155 (DC50 values are 0.31 and 0.76 μM, respectively). STING Degrader-2 can be used to study STING-related autoimmune diseases .
|
-

- HY-156681
-
|
STX-478
|
PI3K
|
Cancer
|
|
STX-478 (compound 80) is an oral CNS-penetrant allosteric mutant-selective PI3Kα inhibitor. STX-478 shows robust and durable tumor regression and can be used in cancer research .
|
-

- HY-177534
-
|
|
MAGL
|
Metabolic Disease
|
|
MGL-IN-2 (Compound 1) is a MGL inhibitor with IC50 values of 0.006 μM and 0.0121 μM for mutant MGL and wild-type MGL, respectively. MGL-IN-2 can be used in the research of metabolic diseases .
|
-

- HY-10542A
-
|
|
Atg8/LC3
Raf
|
Neurological Disease
|
|
(Z)-GW 5074 is a compound which interacts with both mHTT (mutant huntingtin protein) and LC3, but not but not with the wild-type HTT protein. (Z)-GW 5074 inhibits c-Raf, shows no effect on autophagy, and is effective for neurodegenerative disorder .
|
-

- HY-176131
-
|
|
Btk
|
Inflammation/Immunology
Cancer
|
|
AMX5160 (36) is an orally active BTK inhibitor, with an IC50 of 0.98 nM against mutant BTK C481S. AMX5160 (36) can be used in the research for leukemia, lymphoma, multiple sclerosis, and other autoimmune diseases .
|
-

- HY-142161
-
ABD957
1 Publications Verification
|
MAGL
|
Cancer
|
|
ABD957 is a potent and selective covalent inhibitor of the ABHD17 family of depalmitoylases, with an IC50 of 0.21 μM for ABHD17B. ABD957 can block N-Ras signaling and the growth of NRAS-mutant AML cells .
|
-

- HY-125199
-
|
|
ULK
|
Cancer
|
|
ULK-100 is a highly selective and potent ULK1 kinase inhibitor with an IC50 value of 1.6 nM. ULK-100 is promising for research of autophagy-related diseases (e.g., KRAS-mutant lung cancer, glioblastoma) .
|
-

- HY-161031
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-93 (compound 18) is an allosteric inhibitor of T790M/L858R double mutant EGFR. EGFR-IN-93 can be used for non-small lung cancer (NSCLC) research .
|
-

- HY-176792
-
|
|
PROTACs
Ras
|
Cancer
|
|
ACBI-4 is a selective GTP-loaded active state of KRAS (KRAS(on)) PROTAC degrader. ACBI-4 has significant anti-proliferative effect and potently degrades multiple KRAS mutants in cancer cells, such as KRASG12R .
|
-

- HY-151938
-
|
|
HIV
|
Infection
|
|
Reverse transcriptase-IN-3 is a pyrimidine-5-carboxamide derivative, acts as an inhibitor of HIV-1. Reverse transcriptase-IN-3 shows potent activity against the HIV-1 wild-type and mutant strains .
|
-

- HY-108639
-
MIRA-1
2 Publications Verification
NSC 19630
|
MDM-2/p53
Apoptosis
|
Cancer
|
|
MIRA-1 is a maleimide analogue. MIRA-1 can induce apoptosis in mutant p53 cells via restoration of p53-dependent transcriptional transactivation. MIRA-1 has anticancer activity .
|
-

- HY-129943
-
|
|
Bacterial
|
Infection
|
|
Benzothiohydrazide is an analogue of anti–tubercular agent Isoniazid. Benzothiohydrazide exhibits anti–tubercular activity, with MICs of 132 μM and 264 μM for M. tuberculosis wild type (H37Rv) and clinical mutant strains (IC1 and IC2) .
|
-

- HY-N6674
-
|
ECO-4601; TLN-4601; BU 4664L
|
Ras
Apoptosis
|
Cancer
|
|
Diazepinomicin (TLN-4601) is a secondary metabolite produced by Micromonospora sp. Diazepinomicin (TLN-4601) inhibits the EGF-induced Ras-ERK MAPK signaling pathway and induces apoptosis. An anti-tumor agent for K-Ras mutant models .
|
-

- HY-126464
-
|
EoM
|
Androgen Receptor
|
Cancer
|
|
Estromustine (EoM) is the active metabolite of Estramustine phosphate (HY-13627A). Estromustine binds to mutant androgen receptor (m-AR) with EC50 of 2.6 μM in LNCaP, exhibits cytotoxicity in human prostate cells LNCaP with IC50 of 9.73 μM .
|
-

- HY-15734
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
AGI-6780 that potently and selectively inhibits the tumor-associated mutant IDH2 R140Q with IC50 of 23±1.7 nM. AGI-6780 is less potent against IDH2 WT with IC50 of 190±8.1 nM.
|
-

- HY-145701
-
|
|
MEK
|
Cancer
|
|
MEK1/2-IN-2 is a potent ATP-competitive MEK1/2 inhibitor and shows equipotent inhibition of WT MEK1/2 and a panel of MEK1/2 mutant cell lines .
|
-

- HY-161602
-
|
|
EGFR
|
Cancer
|
|
EGFR/HER2-IN-13 (Compd 38) is an inhibitor of mutant EGFR/HER2s which are resistant to other drugs. EGFR/HER2-IN-13 can be used for cancer research .
|
-

- HY-170019
-
|
|
RET
|
Cancer
|
|
RET-IN-28 (Compound 16) is a RET (transmembrane receptor tyrosine protein kinase) inhibitor. RET-IN-28 inhibits the activity of the mutant RET enzyme (RET-V804M), and can be applied to cancer research .
|
-

- HY-164464
-
|
|
EGFR
|
Cancer
|
|
BPI-15086 an orally active, potent, irreversible mutant-selective inhibitor of both EGFR and T790M resistance mutations tyrosine kinase. BPI-15086 can be used for the research of non-small-cell lung cancer .
|
-

- HY-161603
-
|
|
EGFR
|
Cancer
|
|
EGFR/HER2-IN-14 (Compd 46) is an inhibitor of mutant EGFR/HER2s which are resistant to other drugs. EGFR/HER2-IN-14 can be used for cancer research .
|
-

- HY-175519
-
|
|
HIV
|
Infection
|
|
NNRT-IN-11 (14l) is a potent non-nucleoside reverse transcriptase (NNRT) inhibitor with EC50 values of 6.50 to 52.9 nM against WT and a panel of HIV-1 mutants. NNRT-IN-11 has antiviral activity .
|
-

- HY-168579
-
|
|
c-Kit
|
Cancer
|
|
c-Kit-IN-6 (Compound 101) is an inhibitor of c-Kit with an IC50 value ranging between 100 nM and 1 μM. Additionally, c-Kit-IN-6 can inhibit the proliferation of GIST430 and BaF3 mutant cells .
|
-

- HY-10159AR
-
|
AMN107 monohydrochloride monohydrate (Standard)
|
Reference Standards
Bcr-Abl
Autophagy
|
Cancer
|
|
Nilotinib (monohydrochloride monohydrate) (Standard) is the analytical standard of Nilotinib (monohydrochloride monohydrate). This product is intended for research and analytical applications. Nilotinib monohydrochloride monohydrate is a second generation tyrosine kinase inhibitor (TKI), is significantly potent against BCR-ABL, and is active against many BCR-ABL mutants.
|
-

- HY-132609
-
|
|
Small Interfering RNA (siRNA)
Transthyretin (TTR)
|
Neurological Disease
|
|
Patisiran sodium is a double-stranded small interfering RNA that targets a sequence within the transthyretin (TTR) messenger RNA. Patisiran sodium specifically inhibits hepatic synthesis of mutant and wild-type TTR. Patisiran sodium can be used for the research of hereditary TTR amyloidosis .
|
-

- HY-173446
-
|
|
PARP
|
Cancer
|
|
PARP1-IN-38 (compound ent-6_P) is a potent PARP1 inhibitor with an IC50 of 10 μM. PARP1-IN-38 shows selective cytotoxic activity in BRCA mutant cancer cells
|
-

- HY-155722
-
|
|
Bacterial
|
Infection
|
|
Mtb-IN-5 (compound (-)17j) is an isoxazole, with anti-Mycobacterium tuberculosis (Mtb) activity. Mtb-IN-4 inhibits Mtb respiration and biofilm formation in macrophage, and enhances antibiotic isoniazid (INH) inhibition against INH-resistant Mtb mutant .
|
-

- HY-150245
-
|
|
Huntingtin
|
Neurological Disease
|
|
mHTT-IN-1 (Example 1) is a potent mutant huntingtin (mHTT) inhibitor. mHTT is toxic and a major cause of the inherited autosomal dominant neurodegenerative disorder, Huntington's disease (HD). mHTT-IN-1 conducts the reduction of mHTT with an EC50 value of 46 nM .
|
-

- HY-12090R
-
|
|
PCSK9
|
Cardiovascular Disease
|
|
Anacetrapib (Standard) is the analytical standard of Anacetrapib. This product is intended for research and analytical applications. Anacetrapib is a potent CETP inhibitor, with IC50s of 7.9±2.5 nM and 11.8±1.9 nM for rhCETP and C13S CETP mutant, respectively.
|
-

- HY-117407
-
|
|
Smo
|
Cancer
|
|
ALLO-2 is a potent drug-resistant Smoothened (Smo) mutant antagonist that inhibits Smo agonist Hh-Ag1.5-induced luciferase expression in TM3-Gli-Luc cells with IC50 of 6 nM .
|
-

- HY-167948
-
|
|
EGFR
|
Cancer
|
|
Dacomitinib metabolite M1/2 is a potent inhibitor of both wild-type (WT) EGFR and the T790M mutation, demonstrating significant activity against acquired resistance mechanisms in EGFR-mutant non-small-cell lung cancer (NSCLC).
|
-

- HY-P5968
-
|
β(25-35)KA
|
Amyloid-β
|
Neurological Disease
|
|
[Ala28]-β Amyloid(25-35) (β(25-35)KA) is an electrically neutral mutant peptide of Aβ(25-35) that accelerates the aggregation of Firefly Luciferase .
|
-
![[Ala28]-β Amyloid(25-35)](//file.medchemexpress.eu/product_pic/hy-p5968.gif)
- HY-173155
-
|
|
Akt
|
Cancer
|
|
AKT1-IN-9 (4) is a selective mutant AKT1(E17K) inhibitor, with EC50 values of 9 nM and 995 nM in LAPC4-CR and SkBr3 cells, respectively .
|
-

- HY-174245
-
|
|
RET
|
Cancer
|
|
RET-IN-30 (Compound 28) is a RET inhibitor that exhibits inhibitory activity against both wild-type RET and some of its mutants. RET-IN-30 can inhibit the proliferation of A549 cells and has anti-tumor activity .
|
-

- HY-139997
-
|
|
PROTACs
EGFR
|
Cancer
|
|
DDC-01-163 is a mutant-selective allosteric PROTAC-based EGFR degrader, with an IC50 of 45 nM (Pink: EGFR ligand (HY-151048); Black: linker (HY-W008005); Blue: CRBN ligand (HY-14658)) .
|
-

- HY-16905
-
|
|
Src
|
Cancer
|
|
T338C Src-IN-1 is a potent mutant-Src T338C inhibitor; exhibited the most potent inhibition of T338C(IC50=111 nM) relative to WT c-Src (10-fold increase).
|
-

- HY-12602
-
|
|
PARP
|
Cancer
|
|
TNKS-IN-4 (compound 49) is an orally active tankyrase (TNKS) inhibitor with IC50 values of 0.1 nM and 7.6nM for TNKS1 and TNKS2, respectively. TNKS-IN-4 can be used for study of APC-mutant colorectal cancer .
|
-

- HY-P99701
-
|
BMS-986004
|
TNF Receptor
|
Inflammation/Immunology
|
|
Letolizumab (BMS-986004) is a monoclonal antibody targeting CD40L, which is produced to express mutant IgG1 lacking effector function, including Fc binding and complement fixation. Letolizumab reduces rejection, thromboembolism and prolongs the survival time .
|
-

- HY-14716
-
|
|
DNA/RNA Synthesis
Raf
|
Cancer
|
|
CCT239065 is an orally available, effective inhibitor of the mutant protein kinase V600EBRAF (RAF) with anti-tumor activity. CCT239065 inhibits downstream signaling of V600EBRAF in cancer cells, blocking DNA synthesis and suppressing proliferation .
|
-

- HY-P10610
-
|
|
MDM-2/p53
|
Cancer
|
|
Peptide 234CM is a peptide containing isoleucine at position 3, corresponding to the sequence of a point mutation in p53 codon 234. Peptide 234CM induces potent cytotoxic T cell (CTL) and antitumor immune responses against mutant p53 .
|
-

- HY-139884
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-18 potently inhibits enzymatic activity in L858R/T790M/C797S mutant EGFR (4.9 nM), with a significantly lower activity for wild-type EGFR (47 nM).
|
-

- HY-P5968A
-
|
β(25-35)KA TFA
|
Amyloid-β
|
Neurological Disease
|
|
[Ala28]-β Amyloid(25-35) (β(25-35)KA) (TFA) is an electrically neutral mutant peptide of Aβ(25-35) that accelerates the aggregation of Firefly Luciferase .
|
-
![[Ala28]-β Amyloid(25-35) TFA](//file.medchemexpress.eu/product_pic/hy-p5968a.gif)
- HY-79077
-
|
AZD-9291 dimesylate; Mereletinib dimesylate
|
EGFR
|
Cancer
|
|
Osimertinib dimesylate (AZD-9291 dimesylate) is an irreversible and mutant selective EGFR inhibitor with IC50s of 12 and 1 nM against EGFR L858R and EGFR L858R/T790M, respectively.
|
-

- HY-164482
-
|
|
RET
|
Cancer
|
|
HG-6-63-01 is a type II RET tyrosine kinase inhibitor (TKI). HG-6-63-01 also inhibits REarranged during Transfection (RET) kinase and signaling in human thyroid cancer cell lines carrying oncogenic RET alleles. HG-6-63-01 impairs phosphorylation and signalling of RET oncogenic mutants. HG-6-63-01 blunts proliferation of RET/C634R and RET/M918T-transformed fibroblasts and of RET mutant thyroid cancer cells, which is promising for research of cancers harboring oncogenic activation of RET .
|
-

- HY-112306R
-
|
|
c-Kit
PDGFR
FLT3
VEGFR
Apoptosis
|
Cancer
|
|
Ripretinib (Standard) is the analytical standard of Ripretinib. This product is intended for research and analytical applications. Ripretinib (DCC-2618) is an orally bioavailable, selective KIT and PDGFRA switch-control inhibitor. Ripretinib (DCC-2618) targets and binds to both wild-type and mutant forms of KIT and PDGFRA specifically at their switch pocket binding sites, thereby preventing the switch from inactive to active conformations of these kinases and inactivating their wild-type and mutant forms. Ripretinib (DCC-2618) also inhibits multiple other kinase targets, such as FLT3 and KDR (or VEGFR-2) . DCC-2618 exerts antineoplastic effect and induces apoptosis .
|
-

- HY-176135
-
|
|
Histone Methyltransferase
|
Cancer
|
|
BBDDL2204 (compound 13) is a potent and selective EZH2 covalent inhibitor. BBDDL2204 inhibits EZH2 Y641F with an IC50 of 2.5 nM .
|
-

- HY-164488
-
|
|
PAK
|
Cancer
|
|
KT D606 is an inhibitor of the PAK kinase family, with an IC50 value of 4 μM. KT D606 selectively blocks the proliferation of cancer cells transformed by oncogenic RAS mutants and can be used for research on RAS/PAK1-induced cancers .
|
-

- HY-15729
-
|
CO-1686; AVL-301; CNX-419
|
EGFR
|
Cancer
|
|
Rociletinib (CO-1686) is an orally delivered kinase inhibitor that specifically targets the mutant forms of EGFR including T790M, and the Ki values for EGFRL858R/T790M and EGFRWT are 21.5 nM and 303.3 nM, respectively.
|
-

- HY-110392
-
|
|
Estrogen Receptor/ERR
|
Cancer
|
|
CMP8, a selective ligand for estrogen receptor, binds to the mutant estrogen receptor ligand binding domain (ERLBD). CMP8 exhibits IC50 values of 29 nM , 41 nM, 1100 nM and 2200 nM for MGERα, MGRERα, hERα and hERβ, respectively .
|
-

- HY-100214
-
|
|
EGFR
|
Cancer
|
|
EAI001 is a potent, selective mutant epidermal growth factor receptor (EGFR) allosteric inhibitor with an IC50 value of 24 nM for EGFR L858R/T790M. EAI001 can be used for research of cancer .
|
-

- HY-144882
-
-

- HY-103682
-
|
|
Histone Methyltransferase
|
Cancer
|
|
PF-06726304 is a potent and selective EZH2 inhibitor. PF-06726304 inhibits wild-type and Y641N mutant EZH2 with Kis of 0.7 and 3.0 nM, respectively. PF-06726304 displays robust antitumor growth activity .
|
-

- HY-145411
-
|
|
Liposome
|
Endocrinology
|
|
PEG2000-C-DMG, a pegylated lipid, can be used for the preparation of Onpattro. Onpattro, a hepatically directed investigational RNAi therapeutic agent, harnesses this process to reduce the production of mutant and wild-type transthyretin by targeting the 3′ untranslated region of transthyretin mRNA .
|
-

- HY-103682A
-
|
|
Histone Methyltransferase
|
Cancer
|
|
PF-06726304 acetate is a potent and selective EZH2 inhibitor. PF-06726304 acetate inhibits wild-type and Y641N mutant EZH2 with Kis of 0.7 and 3.0 nM, respectively. PF-06726304 acetate displays robust antitumor growth activity .
|
-

- HY-P10610A
-
|
|
MDM-2/p53
|
Cancer
|
|
Peptide 234CM TFA is a peptide containing isoleucine at position 3, corresponding to the sequence of a point mutation in p53 codon 234. Peptide 234CM TFA induces potent cytotoxic T cell (CTL) and antitumor immune responses against mutant p53 .
|
-

- HY-145468
-
|
|
GPR52
|
Neurological Disease
|
|
GPR52 antagonist-1 (Compound 43) is a GPR52 antagonist with an IC50 of 0.63 μM. GPR52 antagonist-1 reduces mHTT (mutant huntingtin protein) levels by targeting GPR52 and promotes survival of mouse primary striatal neurons .
|
-

- HY-168581
-
|
|
c-Kit
|
Cancer
|
|
c-Kit-IN-8 (Compound 53) is an inhibitor for c-Kit kinase, that inhibits uKIT kinase with an IC50 > 1 μM. c-Kit-IN-8 inhibits the proliferation of cancer cell GIST430 and BaF3 wildtype and mutants with IC50 > 0.1 μM .
|
-

- HY-126143
-
|
|
Bcr-Abl
|
Cancer
|
|
CHMFL-ABL-039 is a type II native ABL kinase and drug-resistant V299L mutant BCR-ABL inhibitor with the IC50s of 7.9 nM and 27.9 nM, respectively. CHMFL-ABL-039 is used in the research of chronic myeloid leukemia .
|
-

- HY-17654
-
|
|
EGFR
mTOR
|
Cancer
|
|
BIEGi-1 is an EGFR inhibitor. BIEGi-1 effectively disrupts the EGFR-Rheb interaction in cells. BIEGi-1 robustly inhibits EGFR kinase activity (reduces p-Y1068-EGFR) as well as mTORC1 activation (reduces p-T389-S6K1) in EGFR-mutant cells. BIEGi-1 shows strong antiproliferative effects on EGFR-mutant PC9 and HCC827 cells with IC50 values of 17 nM and 20 nM, respectively. BIEGi-1 can be used for the study of cancers harboring EGFR mutations, such as non-small cell lung cancer (NSCLC) .
|
-

- HY-19851
-
|
GS-9131
|
Reverse Transcriptase
HIV
|
Cancer
|
|
Rovafovir etalafenamide (GS-9131), a proagent of the adenosine nucleotide analogue GS-9148, is an orally active nucleoside reverse transcriptase inhibitor (NRTI). Rovafovir etalafenamide is potent and active against a variety of NRTI mutants, and shows potent anti-HIV-1 activity .
|
-

- HY-159163
-
|
|
Ras
|
Cancer
|
|
KRASG12C IN-12 (compound-1) is a KRAS G12C inhibitor. KRASG12C IN-12 (compound-1) can form a ternary complex with intracellular CYPA and the activated KRAS G12C mutant .
|
-

- HY-172582
-
|
|
CDK
|
Cancer
|
|
CDK9-IN-38 (compound 14) is a CDK9 inhibitor with IC50 values of 1.2 nM and 3.3 nM for CDK9 wild-type and L156F mutant, respectively. CDK9-IN-38 inhibits tumor growth in vivo and in vitro .
|
-

- HY-B0282S
-
|
ACh-d4 chloride
|
nAChR
Calcium Channel
Endogenous Metabolite
|
Neurological Disease
Cancer
|
|
Acetylcholine-d4 (chloride) is the deuterium labeled Acetylcholine chloride. Acetylcholine chloride (ACh chloride), a neurotransmitter, is a potent cholinergic agonist. Acetylcholine chloride is a modulator of the activity of dopaminergic (DAergic) neurons through the stimulation of nicotinic acetylcholine receptors (nAChRs) . Acetylcholine chloride inhibits p53 mutant peptide aggregation in vitro .
|
-

- HY-15729A
-
|
CO-1686 hydrobromide; AVL-301 hydrobromide; CNX-419 hydrobromide
|
EGFR
|
Cancer
|
|
Rociletinib hydrobromide (CO-1686 hydrobromide) is an orally delivered kinase inhibitor that specifically targets the mutant forms of EGFR including T790M, and the Ki values for EGFRL858R/T790M and EGFRWT are 21.5 nM and 303.3 nM, respectively.
|
-

- HY-177453
-
|
DYNE-101 oligomer
|
Antibody-Oligonucleotide Conjugates (AOCs)
|
Others
|
|
Basivarsen (DYNE-101 oligomer) is composed of an antisense oligonucleotides and a linker (HY-177454). It is designed to target mutant nuclear myotonic dystrophy protein kinase (DMPK) RNA for RHase H-mediated degradation to correct splicing. It is used for the study of myotonic dystrophy type 1 (DM1).
|
-

- HY-146145
-
|
|
Fungal
Mitochondrial Metabolism
|
Infection
|
|
Metyltetraprole is a promising fungicide with EC50 values of both 0.002 ppm against sensitive wild-type and G143A mutant of Zymoseptoria tritici. Metyltetraprole is effective against QoI (quinone outside inhibitor) resistant strains. Metyltetraprole inhibits the respiratory chain via complex III .
|
-

- HY-N0328
-
-

- HY-168512
-
|
|
Raf
|
Cancer
|
|
BRAFV600E-IN-1 (compound 9S) is a BRAF inhibitor. BRAFV600E-IN-1 has a significant apoptosis-inducing effect in cell lines expressing mutant KRAS and cancer cells expressing BRAFV600E .
|
-

- HY-16767
-
|
MK-1439
|
HIV
Reverse Transcriptase
|
Infection
|
|
Doravirine (MK-1439) is a highly specific HIV-1 nonnucleoside reverse transcriptase inhibitor with IC50s of 4.5 nM, 5.5 nM and 6.1 nM against the wild type and K103N and Y181C reverse transcriptase mutants, respectively .
|
-

- HY-157808
-
|
|
Bacterial
|
Infection
|
|
Antibacterial agent 185 (compound IP-01) is a potent antibacterial agent. Antibacterial agent 185 inhibits filamentous temperature-sensitive mutant Z (FtsZ) polymerization and bundling by increasing GTP hydrolysis. Antibacterial agent 185 inhibits Streptococcus pneumoniae and shows narrow-spectrum activity .
|
-

- HY-179300A
-
|
|
Ras
|
Cancer
|
|
KRAS-IN-48 (Compound 1-01) is a KRAS mutant inhibitor, with Kd values of 2.58 nM and 5.49 μM for KRAS-G12D and KRAS-G12V, respectively. KRAS-IN-48 can be used in the research of cancer .
|
-

- HY-B0282S1
-
|
ACh-d9(chloride)
|
nAChR
Calcium Channel
Endogenous Metabolite
|
Neurological Disease
Cancer
|
|
Acetylcholine-d9 (chloride) is the deuterium labeled Acetylcholine chloride. Acetylcholine chloride (ACh chloride), a neurotransmitter, is a potent cholinergic agonist. Acetylcholine chloride is a modulator of the activity of dopaminergic (DAergic) neurons through the stimulation of nicotinic acetylcholine receptors (nAChRs) . Acetylcholine chloride inhibits p53 mutant peptide aggregation in vitro .
|
-

- HY-114604
-
|
|
Bacterial
|
Others
|
|
Propioxatin B is a tricyclic sesquiterpenoid compound isolated from the root of vetiver grass. It has anti-tuberculosis activity and inhibitory effects on a variety of drug-resistant mutants of Mycobacterium tuberculosis. In computer simulation docking studies, it showed binding affinity with bacterial DNA gyrase and has a certain safety in vivo.
|
-

- HY-143734
-
|
|
EGFR
|
Cancer
|
|
HER2-IN-6 is a potent inhibitor of HER2. HER2-IN-6 has the potential for the research of wild and/or mutant EGFR and/or HER2 kinase mediated tumor (extracted from patent WO2021164697A1, compound 11) .
|
-

- HY-12343
-
|
CID-53347902
|
Potassium Channel
|
Cardiovascular Disease
|
|
ML277 (CID-53347902) is a potent and selective activator of K(v)7.1 (KCNQ1) potassium channel activator (EC50=270 nM), rescues function of pathophysiologically important mutant channel complexes in human induced pluripotent stem cell-derived cardiomyocytes .
|
-

- HY-W287494
-
|
|
MDM-2/p53
|
Cancer
|
|
Antitumor agent-202 is a stabilizer of the p53 Y220C mutant. It exhibits a selective inhibitory effect on the proliferation of tumor cells carrying the p53 Y220C mutation and can be applied to the research of cancers associated with the p53 Y220C mutation .
|
-

- HY-126362
-
|
|
Glycosidase
|
Others
|
|
ML266 is glucocerebrosidase (GCase) molecule chaperone with IC50 of 2.5 µM. ML266 binds to GCase and transports of the mutant protein to the lysosome, and resume the activity of GCase. ML266 dose not inhibit the GCase enzyme’s action. ML266 has the potential for the research of gaucher disease .
|
-

- HY-122045
-
-

- HY-163462
-
|
|
Fungal
|
Infection
|
|
Poacic Acid is a plant-derived stilbenoid with an antifungal activity. Poacic Acid localizes to the yeast cell wall and disrupts the production and assembly of β-1,3-glucan in the fungal cell walls. Poacic Acid exhibits fungicidal activity to Saccharomyces cerevisiae and plasma membrane-compromised Candida albicans mutants .
|
-

- HY-153951S
-
|
|
Btk
|
Inflammation/Immunology
Cancer
|
|
BTK-IN-26 (compound 18) is a potent inhibitor of Bruton's tyrosine kinase (BTK) and its C481 mutant, with IC50 values of 0.7 and 0.8 nM for BTK and BTK C481S, respectively. BTK-IN-26 can be used for cancer and autoimmune diseases research .
|
-

- HY-154959A
-
|
|
Ras
|
Cancer
|
|
(9R,12aR)-AZD4747 is a diastereomer of AZD4747 (HY-154959). AZD4747 is a selective mutant GTPase KRAS G12C inhibitor with blood-brain barrier permeability. AZD4747 has the potential to study cancer .
|
-

- HY-B0248
-
|
|
Glucocorticoid Receptor
Huntingtin
Apoptosis
|
Neurological Disease
Inflammation/Immunology
|
|
Desonide is a non-fluorinated corticosteroid anti-inflammatory agent that acts on the glucocorticoid receptor. Desonide can also specifically bind to the mutant huntingtin protein (mHTT), reducing the level and toxicity of mHTT. Desonide can be used in the research of Huntington's disease and inflammatory diseases such as atopic dermatitis .
|
-

- HY-144605
-
|
|
EGFR
PROTACs
|
Cancer
|
|
PROTAC EGFR degrader 3 is a potent PROTAC EGFR degrader. PROTAC EGFR degrader 3 shows excellent cellular activity against the H1975 and HCC827 cells with high selectively. PROTAC EGFR degrader 3 shows that the lysosome is involved in the degradation process of EGFR mutant degradation .
|
-

- HY-P99823
-
|
TA-46; sFGFR3
|
FGFR
|
Others
|
|
Recifercept (TA-46) is a soluble, recombinant fibroblast growth factor receptor 3 (FGFR3) molecule. Recifercept can be used as a decoy/ligand trap to decrease the amount of fibroblast growth factors that can bind to mutant FGFR3 receptors. Recifercept can be used for the research of achondroplasia .
|
-

- HY-153609
-
|
|
Transthyretin (TTR)
Small Interfering RNA (siRNA)
|
Neurological Disease
|
|
AS-Patisiran sodium is an antisense strand of Patisiran. Patisiran is a double-stranded small interfering RNA that targets a sequence within the transthyretin (TTR) messenger RNA. Patisiran specifically inhibits hepatic synthesis of mutant and wild-type TTR. Patisiran can be used for the research of hereditary TTR amyloidosis .
|
-

- HY-18690R
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Enasidenib (Standard) is the analytical standard of Enasidenib. This product is intended for research and analytical applications. Enasidenib is an oral, potent, reversible, selective inhibitor of the IDH2 mutant enzymes, with IC50s of 100 and 400 nM against IDH2R140Q and IDH2R172K, respectively.
|
-

- HY-18606
-
|
|
Ras
|
Cancer
|
|
K-Ras G12C-IN-3 (Compound VI-5) is an irreversible small molecule inhibitor of mutant K-Ras G12C. K-Ras G12C-IN-3 can be used in the research of cancers .
|
-

- HY-151211
-
|
|
Btk
|
Inflammation/Immunology
Cancer
|
|
BTK-IN-16 is a dual inhibitor of BTK wild type and C481S mutant of Bruton’s tyrosine kinase (BTK) with IC50s of 5.1 and 4.1 μM. BTK-IN-16 can be used for the research of autoimmune diseases and chronic lymphocytic leukemia .
|
-

- HY-113646
-
|
|
FKBP
|
Others
|
|
Shield-2 is an efficient stabilizing ligand binding to the FKBP (F36V) protein with a dissociation constant of 29 nM. Shield-2 binds tightly to the FKBP mutants destabilizing domains and prevents degradation, thus providing small molecule regulation over intracellular protein levels .
|
-

- HY-121736
-
|
AGI-026
|
Isocitrate Dehydrogenase (IDH)
|
Neurological Disease
|
|
AGI-12026 is brain-penetrant dual inhibitor of mutant IDH1 and 2. AGI-12026 shows partial inhibition of the IDH1-R132H homodimer as allosteric modulators. AGI-12026 has the potential for research of glioma .
|
-

- HY-152161
-
|
|
HIV
Reverse Transcriptase
|
Infection
|
|
HIV-1 inhibitor-51, a non-nucleoside reverse transcriptase inhibitor (NNRTI), exhibits outstanding antiviral activity against WT HIV-1 (IIIB) and a panel of mutant strains. HIV-1 inhibitor-51 has high binding affinity (KD=2.50 μM) and inhibitory activity (IC50=0.03 μM) to WT HIV-1 RT. HIV-1 inhibitor-51 has EC50s of 2.22-53.3 nM for mutant strains (L100I, K103N, Y181C, Y188L, E138K, F227L + V106A, RES056) .
|
-
- HY-178497
-
|
|
PROTACs
Ras
p38 MAPK
TNF Receptor
|
Inflammation/Immunology
Cancer
|
|
ZJK-807 is a highly effective and selective PROTAC degrader targeting KRASG12D (DC50 = 79.5 nM in AsPC-1 cells). ZJK-807 shows minimal impact on wild-type KRAS or other mutants (G12C/S/V, G13D), inducing mutant-specific cytotoxicity. ZJK-807 suppresses RAS/MAPK signaling and uniquely modulates TNF signaling and eukaryotic ribosome biogenesis. ZJK-807 can be used for the study of KRAS-driven pancreatic cancer. Yellow: KRASG12D ligand (HY-W087383); Green: E3 ligase CRBN ligand (HY-178507); Black: Linker (HY-178506) .
|
-

- HY-115443
-
|
|
FLT3
|
Cancer
|
|
MZH29 is a potent and orally active FLT3 inhibitor. MZH29 shows inhibitory effects on wild-type and mutant FLT3, including the FLT3-ITD, FLT3-D835H/Y/V and FLT3-K663Q mutants. MZH29 retains its potent inhibitory effect against the FLT3-ITD/F691L mutation, a drug resistance mutation against the well-known FLT3 inhibitor, AC220 (HY-13001). MZH29 can be used for the research of cancer, such as acute myeloid leukemia (AML) .
|
-

- HY-175491
-
|
|
HIV
Reverse Transcriptase
|
Infection
|
|
HIV-1-IN-85 is an orally active HIV-1 inhibitor (IC50 = 72 nM). HIV-1-IN-85 exhibits strong inhibitory activity against wild-type (WT) HIV-1 and NNRTI-resistant single mutant strains (L100I, K103N, Y181C, Y188L, E138K) and moderate efficacy against double mutant strains (F227L+V106A, RES056). HIV-1-IN-85 shows good in vivo safety in ICR mice. HIV-1-IN-85 can be used for the study of HIV-1 infection .
|
-

- HY-132844
-
|
HL-085
|
MEK
|
Cancer
|
|
Tunlametinib is a highly selective, orally active MEK1/2 inhibitor (IC50=1.9 nM, MEK1). Tunlametinib blocks the RAS-RAF-MEK-ERK signaling pathway, arrests tumor cell cycle and promotes apoptosis. Tunlametinib potently inhibits the proliferation of RAS/RAF mutant cancer cells (such as BRAF V600E, KRAS G12C mutant cells). Tunlametinib shows synergistic anti-tumor effects with BRAF/KRAS G12C/SHP2 inhibitors, Docetaxel (HY-B0011). Tunlametinib can be used to study targeted therapy for RAS/RAF mutation-driven malignancies (such as melanoma, colorectal cancer, and non-small cell lung cancer) .
|
-

- HY-179094
-
|
|
PROTACs
IRAK
NF-κB
Enolase
|
Inflammation/Immunology
Cancer
|
|
PSP-0119 is a highly efficient and effective PROTAC degrader targeting IRAK4 (IC50 = 2.83 nM). PSP-0119 can inhibit IRAK4 kinase activity, NF-κβ activity, and IL-1β-induced IRAK4 phosphorylation. PSP-0119 degrades IRAK4 in FLT3-mutant AML cell lines, sparing FLT3-wild-type AML cells, FLT3-wild-type samples, and normal bone marrow. PSP-0119 downregulates alpha-enolase (eNOS) of MOLM-13 cells. PSP-0119 can be used for the study of Acute Myeloid Leukemia (AML) .
|
-

- HY-E70219
-
|
|
Biochemical Assay Reagents
|
Others
|
|
SpCas9 D10A Nickase is a mutant of the Cas9 protein. SpCas9 D10A Nickase retains the function of a cleavage domain of Cas9 nuclease and specifically cleaves the target single strand to form a nick. SpCas9 D10A Nickase reduces off-target effects .
|
-

- HY-160678
-
|
|
Bacterial
|
Infection
|
|
InhA-IN-7 (Compound 11) is a Triclocan (HY-B1119) derivative with inhibitory activity towards enoyl-acyl carrier protein reductase (InhA) with an IC50 of 96 nM. InhA-IN-7 inhibits proliferations of Mycobacterium tuberculosis wildtype and mutant strains with MICs ranging from 19 to 75 μM .
|
-

- HY-P4528
-
|
|
Ser/Thr Protease
|
Others
|
|
Ala-Phe-Pro-βNA is the subsrate of prolyl tripeptidyl aminopeptidase, which is from Porphyromonas gingivalis. Ala-Phe-Pro-βNA binds to H-Ala-Ile-pyrrolidin-2-yl boronic acid for PTP39 and the E636A mutant with Ki values of 88.1 nM and 48.8 nM, respectivley .
|
-

- HY-175419
-
|
trans bis-Isatoic anhydride
|
DNA/RNA Synthesis
|
Cancer
|
|
TBIA (trans bis-isatoic anhydride) is a covalent RNA crosslinker. TBIA selectively induces RNA tertiary interactions (e.g., multi-helix junctions, loop-helix packing). TBIA is promising for research of RNA higher-order structure and disease-associated RNAs (e.g., KRAS-mutant RNAs) .
|
-

- HY-164513
-
|
|
Ras
Phosphodiesterase (PDE)
|
Cancer
|
|
NHTD is a KRAS-PDEδ inhibitor. NHTD targets the prenyl-binding pocket of PDEδ, altering the cellular localization of KRAS, thereby inhibiting the proliferation of KRAS-mutant cancer cells and inducing apoptosis. NHTD can be used for research on KRAS-driven non-small cell lung cancer (NSCLC) .
|
-

- HY-155720
-
|
|
Bacterial
|
Infection
|
|
Mtb-IN-4 (compound 17h) is a nontoxic isoxazole, with anti-Mycobacterium tuberculosis (Mtb) activity (IC50=0.70 μM). Mtb-IN-4 inhibits Mtb respiration and biofilm formation in macrophage, and enhances antibiotic isoniazid (INH) inhibition against INH-resistant Mtb mutant .
|
-

- HY-122958
-
|
|
α-synuclein
|
Neurological Disease
|
|
Peucedanocoumarin III is an inhibitor of α-synuclein and Huntington protein aggregates that enhances the clearance of nuclear and cytoplasmic β23 aggregates and prevents cytotoxicity induced by disease-associated proteins (i.e., mutant Huntington proteins and α-synuclein). Peucedanocoumarin III may be used in Parkinson's disease research .
|
-

- HY-W127705
-
|
|
Fluorescent Dye
|
Others
|
|
Quinacrine mustard dihydrochloride is a fluorochrome. Quinacrine mustard dihydrochloride as a polycyclic aromatic agent can be used as mutagenic agent induces the mutants of bacteria. Quinacrine mustard dihydrochloride induces cell cycle arrest at G2/M-phase. Quinacrine mustard dihydrochloride has the potential for the research of plant, animal, or human chromosomes .
|
-

- HY-B0282R
-
|
ACh chloride (Standard)
|
Reference Standards
nAChR
Calcium Channel
Endogenous Metabolite
|
Neurological Disease
Cancer
|
|
Acetylcholine (chloride) (Standard) is the analytical standard of Acetylcholine (chloride). This product is intended for research and analytical applications. Acetylcholine chloride (ACh chloride), a neurotransmitter, is a potent cholinergic agonist. Acetylcholine chloride is a modulator of the activity of dopaminergic (DAergic) neurons through the stimulation of nicotinic acetylcholine receptors (nAChRs) . Acetylcholine chloride inhibits p53 mutant peptide aggregation in vitro .
|
-

- HY-18213
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-9 (Compound 8) is a potent EGFR kinase inhibitor with IC50s of 7 nM, 28 nM for the wild type EGFR kinase and double mutant EGFR kinase (L858R/T790M). EGFR-IN-9 has antitumor activity .
|
-

- HY-I0960
-
Uracil
4 Publications Verification
|
Endogenous Metabolite
Drug Derivative
|
Cancer
|
|
Uracil is a pyrimidine derivative and one of the four nucleobases in the nucleic acid of RNA. Uracil is a frequently occurring DNA base damage that results from the spontaneous or chemically induced deamination of cytosine and is mutagenic at the level of replication. Uracil could potentially alter transcriptional fidelity, resulting in production of mutant proteins .
|
-

- HY-114277A
-
|
AMG-510 racemate
|
Ras
p38 MAPK
|
Cancer
|
|
Sotorasib racemate (Compound A) is an orally active racemate of Sotorasib (HY-114277), a covalent inhibitor of KRAS G12C mutant which induces adaptive feedback activation of MAPK pathway. Sotorasib racemate also exerts inhibitor activity against KRAS G12C induced cancer and can be applied to cancer research .
|
-

- HY-13223R
-
|
|
FLT3
PDGFR
Autophagy
|
Cancer
|
|
Crenolanib (Standard) is the analytical standard of Crenolanib. This product is intended for research and analytical applications. Crenolanib is a potent and selective inhibitor of wild-type and mutant isoforms of the class III receptor tyrosine kinases FLT3 and PDGFRα/β with Kds of 0.74 nM and 2.1 nM/3.2 nM, respectively.
|
-

- HY-142517
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-25 is a potent EGFR inhibitor with IC50s of 9 nM and 60 nM for BaF3 cells (EGFR DEL19/T790M/C797S) and A431 cells (WT), respectively .
|
-

- HY-135864
-
|
|
Ras
|
Cancer
|
|
KRAS inhibitor-6 is a potent KRAS G12C inhibitor, extracted from patent WO2017087528A1, compound A .
|
-

- HY-135866
-
|
|
Ras
|
Cancer
|
|
KRAS inhibitor-8 is a potent KRAS G12C inhibitor, extracted from patent WO2017087528A1, compound C .
|
-

- HY-132979
-
|
|
Ras
|
Cancer
|
|
KRAS G12C inhibitor 18 is a potent and orally active KRAS G12C inhibitor. Anti-tumor activities .
|
-

- HY-141450
-
|
|
β-catenin
|
Cancer
|
|
NRX-103095 is an enhancer of the interaction between β-catenin, and its cognate E3 ligase, SCF β-TrCP. NRX-103095 enhances the binding of pSer33/Ser37 β-catenin peptide for β-TrCP with an EC50 of 163 nM .
|
-

- HY-142519
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-27 is a potent EGFR inhibitor with IC50s of <50 nM for EGFR Del, L858R, Del/T790M, L858R/T790M, Del/T790M/C797S, and L858R/T790M/C797S, respectively (WO2021249324A1, compound 511) .
|
-

- HY-168112
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-127 is an ATP-competitive EGFR inhibitor with IC50s of 136.3 nM and 161. 2 nM for EGFR del19 and EGFR del19/T790M/C797S, respectively. EGFR-IN-127 has the potential for the study of non-small-cell lung cancer (NSCLC) .
|
-

- HY-135865
-
|
|
Ras
|
Cancer
|
|
KRAS inhibitor-7 is a potent KRAS G12C inhibitor, extracted from patent WO2017087528A1, compound B .
|
-

- HY-168055
-
|
|
Ligands for E3 Ligase
|
Cancer
|
|
(S)-Deoxy-thalidomide is an E3 ubiquitin ligase ligand. (S)-Deoxy-thalidomide can be linked to the target protein ligand through a linker to form PROTAC K-Ras Degrader-3 (HY-168054). PROTAC K-Ras Degrader-3 can target KRAS mutant proteins for degradation .
|
-

- HY-157287
-
|
|
Others
|
Inflammation/Immunology
|
|
(S)-Phe-A110/B319 is a selective binde that has 20-fold higher affinity towards the H1047R mutant of p110α in the p110α/p85α PI3K complex .
|
-

- HY-110094
-
BIMU 8
1 Publications Verification
|
5-HT Receptor
|
Neurological Disease
|
|
BIMU 8 is a potent and selective 5-HT4 agonist with EC50s of 18 nM, 77 nM, and 540 nM for wild type 5HT4 receptor, T3.36A, and W6.48A mutant 5-HT4 receptors .
|
-

- HY-59219
-
|
|
Drug Intermediate
|
Cancer
|
|
Cyclopropylboronic acid is an intermediate. Cyclopropylboronic acid can be used to synthesize Compound 25. Compound 25 has antiproliferative effects on EGFR mutant (EGFR Δ19del/T790M/C797S) cells. Cyclopropylboronic acid can be used in lung cancer research .
|
-

- HY-151999
-
|
|
Ras
|
Cancer
|
|
KRAS G12C inhibitor 65 is a potent and covalent KRAS G12C inhibitor that traps KRAS G12C in the GDP-bound state. KRASG12C IN-1 exhibits potent antitumor activity against KRAS-mutant non-small cell lung cancer .
|
-

- HY-114956
-
|
|
HBV
|
Infection
|
|
AT-61 is a non nucleoside HBV replication inhibitor. AT-61 prevents the capsid formation of pre genomic RNA, resulting in the production of empty capsids. AT-61 has the activity of drug-resistant mutant strains. AT-61 can be used for research on hepatitis B virus infection .
|
-

- HY-N7484
-
|
|
Fungal
|
Cancer
|
|
(−)-Voacangarine is an indole alkaloid, which exhibits cytotoxic effects against cancer cells HepG2, A375, MDA-MB-231, SH-SY5Y, and CT26 with IC50 of 5~20 mg/mL. (−)-Voacangarine inhibits the cultivation of Saccharomyces cerevisiae wildtype and repair-deficient mutants .
|
-

- HY-135892
-
|
|
MAP4K
|
Inflammation/Immunology
|
|
GNE-1858 is a potent and ATP-competitive hematopoietic progenitor kinase-1 (HPK1) inhibitor, with IC50s of 1.9 nM, 1.9 nM, and 4.5 nM for wild-type and the active mimetic mutants HPK1-TSEE and HPK1-SA, respectively .
|
-

- HY-139047
-
|
|
GLUT
|
Cancer
|
|
SW157765 is a selective non-canonical glucose transporter GLUT8 (SLC2A8) inhibitor. KRAS/KEAP1 double mutant NSCLC cells are selectively sensitive to the SW157765, due to the convergent consequences of dual KRAS and NRF2 modulation of metabolic and xenobiotic gene regulatory programs .
|
-

- HY-114043
-
|
|
Glycosidase
|
Metabolic Disease
|
|
NCGC00092410 is a potent, selective, and nonsugar glucocerebrosidase (GC) inhibitor, with an IC50 of 31 nM. NCGC00092410 shows no activity against the related hydrolases at concentrations up to 77 μM. NCGC00092410, a GC chaperone, and increases the activity and lysosomal localization of glucocerebrosidase in mutant cell lines. NCGC00092410 can be used for the research of Gaucher disease .
|
-

- HY-14780
-
|
NXL 101
|
Antibiotic
Topoisomerase
DNA/RNA Synthesis
Bacterial
|
Infection
|
|
Viquidacin (NXL 101) is an antibiotic with inhibitory activity against topoisomerase IV and DNA gyrase. Viquidacin exhibits antibacterial activity against gram positive bacterial by inhibiting the supercoiling, decatenation and relaxation in strains Staphylococcus aureus and Escherichia coli in micromolar levels. Viquidacin inhibits S. aureus wildtype and mutants with MIC of 2-128 mg/L .
|
-

- HY-168215
-
|
|
PROTACs
Epigenetic Reader Domain
|
Cancer
|
|
YDR1 is a potent PROTAC SMARCA2 degrader, with the DC50 of 7.7 nM. YDR1 plays an important role in SMARCA4 mutant cancers(Sturcture Note:(Blue: Cereblon ligand (HY-W087383), Black: linker;Pink: SMARCA2 ligand (HY-44012B)) .
|
-

- HY-172251
-
|
|
FGFR
Apoptosis
|
Cancer
|
|
FGFR-IN-17 (Compound 12l) is an orally active irreversible inhibitor of FGFR, which has a strong inhibitory effect on FGFR and its mutants. FGFR-IN-17 can inhibit the proliferation and migration, and induce apoptosis of non-small cell lung cancer cells. FGFR-IN-17 has anti-tumor activity .
|
-

- HY-123921
-
|
|
PROTACs
EGFR
|
Cancer
|
|
Gefitinib-based PROTAC 3, conjugating an EGFR binding element to a von Hippel-Lindau ligand via a linker, induces EGFR degradation with DC50s of 11.7 nM and 22.3 nM in HCC827(exon 19 del) and H3255 (L858R mutantion) cells, respectively .
|
-

- HY-160084
-
|
|
DNA Methyltransferase
|
Cancer
|
|
2′-Deoxy-4′-thiocytidine (T-dCyd) is a novel epigenetic agent. 2′-Deoxy-4′-thiocytidine administrations led to a significant p21 increase. 2′-Deoxy-4′-thiocytidine can eradicate of tumor cells in the double-mutant xenografts .
|
-

- HY-W740378
-
|
AFN-941
|
EGFR
JAK
|
Others
|
|
1,2,3,4-Tetrahydrostaurosporine is a derivative of Staurosporine (HY-15141) and an inhibitor of mutant EGFR (IC50=74 nM for EGFRT790M). It is selective for EGFRT790M over wild-type EGFR (IC50=390 nM). It also binds to Janus kinase 3 (JAK3).
|
-

- HY-N12390
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Alternaphenol B2 is a selective IDH1 inhibitor from the coral-derived fungus Parengyodontium album SCSIO SX7W11. Alternaphenol B2 shows inhibitory activity against isocitrate dehydrogenase mutant R132H (IDH1m), with IC50 values of 41.9 μM .
|
-

- HY-125540
-
|
|
Influenza Virus
|
Infection
Inflammation/Immunology
|
|
Saussureamine C shows gastroprotective effect on acidified ethanol-induced gastric mucosal lesions in rats. Saussureamine C inhibits H274Y and N294S mutants and can be used for research of influenza virus infection. Saussureamine C can be isolated from the dried roots of Saussurea lappa Clarke .
|
-

- HY-123766
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-99 (compound 1a) is a potent EGFR and HER2 Exon 20 insertion mutant inhibitor. EGFR-IN-99 has excellent antiproliferative activity against DFCI127 cells, with an EC50 of 11.5 nM. EGFR-IN-99 can be used for the research of non-small cell lung cancer (NSCLC) .
|
-

- HY-101494
-
|
LY3214996
|
ERK
|
Cancer
|
|
Temuterkib (LY3214996) is a highly selective inhibitor of ERK1 and ERK2, with IC50 of 5 nM for both enzymes in biochemical assays. Temuterkib potently inhibits cellular p-RSK1 in BRAF and RAS mutant cancer cell lines. Temuterkib shows potent antitumor activities in cancer models with MAPK pathway alterations.
|
-

- HY-163522
-
|
|
Influenza Virus
|
Infection
|
|
Neuraminidase-IN-20 (Compound 5i) is a potent inhibitor of mutant neuraminidase (NA) (H5N1-H274Y) (IC50= 0.44 μM).Neuraminidase-IN-20 binds to the 430 cavity site of NA and disrupts the enzymatic activity of NA .
|
-

- HY-10638
-
|
|
c-Kit
Apoptosis
|
Cancer
|
|
AP23464 is an ATP-based inhibitor for Kit, that inhibits the phosphorylation of Kit wildtype and mutants, with IC50 of 5-85 nM. AP23464 inhibits the proliferation of Kit mutated cells (IC50 is 3-20 nM), arrests the cell cycle at G0/G1 phase, and induces apoptosis in Kit mutated cells .
|
-

- HY-126287
-
|
|
Trk Receptor
Apoptosis
|
Cancer
|
JND4135 is a Type II TRK inhibitor with IC50 values of 2.79, 3.19, and 3.01 nM against TRKA, TRKB, TRKC, respectively. JND4135 can overcome resistance from TRK xDFG and other mutant forms in the BaF3 stable model, inhibiting phosphorylation of both WT and xDFG mutant TRKs, along with their downstream signaling molecules. JND4135 can induce G0/G1 phase arrest and apoptosis in BaF3–CD74-TRKA-G667C cells. JND4135 shows tumor growth inhibition activity in the BaF3-CD74-TRKA-G667C mouse xenograft model .
|
-

- HY-147183
-
|
|
EGFR
|
Cancer
|
|
JBJ-09-063 is a mutant-selective allosteric EGFR inhibitor with IC50s of 0.147 nM, 0.063 nM, 0.083 nM and 0.396 nM for EGFR L858R, EGFR L858R/T790M, EGFR L858R/T790M/C797S and EGFRLT/L747S. JBJ-09-063 effectively reduces EGFR, Akt and ERK1/2 phosphorylation. JBJ-09-063 is effective across EGFR tyrosine kinase inhibitor (TKI)-sensitive and resistant models. JBJ-09-063 can be used for researching EGFR-mutant lung cancer .
|
-

- HY-147183A
-
|
|
EGFR
|
Cancer
|
|
JBJ-09-063 TFA is a mutant-selective allosteric EGFR inhibitor with IC50s of 0.147 nM, 0.063 nM, 0.083 nM and 0.396 nM for EGFR L858R, EGFR L858R/T790M, EGFR L858R/T790M/C797S and EGFRLT/L747S. JBJ-09-063 TFA effectively reduces EGFR, Akt and ERK1/2 phosphorylation. JBJ-09-063 TFA is effective across EGFR tyrosine kinase inhibitor (TKI)-sensitive and resistant models. JBJ-09-063 TFA can be used for researching EGFR-mutant lung cancer .
|
-

- HY-147183B
-
|
|
EGFR
|
Cancer
|
|
JBJ-09-063 hydrochloride is a mutant-selective allosteric EGFR inhibitor with IC50s of 0.147 nM, 0.063 nM, 0.083 nM and 0.396 nM for EGFR L858R, EGFR L858R/T790M, EGFR L858R/T790M/C797S and EGFRLT/L747S. JBJ-09-063 hydrochloride effectively reduces EGFR, Akt and ERK1/2 phosphorylation. JBJ-09-063 hydrochloride is effective across EGFR tyrosine kinase inhibitor (TKI)-sensitive and resistant models. JBJ-09-063 hydrochloride can be used for researching EGFR-mutant lung cancer .
|
-

- HY-18767
-
|
AG-120
|
Isocitrate Dehydrogenase (IDH)
|
Metabolic Disease
Cancer
|
|
Ivosidenib (AG-120) is an orally active inhibitor of isocitrate dehydrogenase 1 mutant (mIDH1) enzyme, it exhibits profound d-2-hydroxyglutatrate (2-HG) lowering in vivo. Ivosidenib (AG-120) has the potential for AML therapy due to its acceptable safety profile and clinical activity .
|
-

- HY-136938
-
NEO2734
3 Publications Verification
EP31670
|
Epigenetic Reader Domain
Histone Acetyltransferase
|
Cancer
|
|
NEO2734 (EP31670) is an orally active dual p300/CBP and BET bromodomain selective inhibitor, with IC50 values of <30 nM for both p300/CBP and BET bromodomains . NEO2734 is active in SPOP mutant and wild-type prostate cancer .
|
-

- HY-130492
-
ARCC-4
2 Publications Verification
|
PROTACs
Androgen Receptor
|
Cancer
|
|
ARCC-4 is a low-nanomolar Androgen Receptor (AR) degrader based on PROTAC, with a DC50 of 5?nM. ARCC-4 is an enzalutamide-based von Hippel-Lindau (VHL)-recruiting AR PROTAC and outperforms enzalutamide. ARCC-4 effectively degrades clinically relevant AR mutants associated with antiandrogen therapy .
|
-

- HY-149928
-
|
|
HIV
|
Infection
|
|
NNRT-IN-1 (compound 8r) is a potent non-nucleoside reverse transcriptase (NNRT) inhibitor featuring significantly anti-resistance efficacy. NNRT-IN-1 inhibits the wild-type HIV-1 and five mutant strains with EC50s in the nanomolar range. NNRT-IN-1 displays favorable pharmacokinetic properties .
|
-

- HY-158039
-
|
|
Deubiquitinase
Apoptosis
|
Cancer
|
|
YCH2823 is an inhibitor of USP7 (IC50 = 49.6 nM; Kd = 0.117 μM). YCH2823 shows significant efficacy in inhibiting TP53 wild-type and mutant tumors, with approximately 5-fold higher potency than FT671. YCH2823 induce apoptosis. YCH2823 synergistic effects with mTOR inhibitors .
|
-

- HY-141523
-
|
RMC-4630; SHP2-IN-7
|
SHP2
Phosphatase
|
Cancer
|
|
Vociprotafib (RMC-4630) is an orally active, selective and potent phosphatase SHP2 inhibitor, which blocks activation of the RAS-RAF-MEK-ERK signaling pathway with antitumor activity. Vociprotafib accelerates the time to, and increases the magnitude of, tumor regressions in Osimertinib (HY-15772)-sensitive EGFR-mutant tumors of mice .
|
-

- HY-164389
-
|
|
Ras
|
Cancer
|
|
SML-10-70-1 is a ligand for RAS, which covalently modifies the K-Ras G12C mutant protein, and inhibits the phosphorylation of ERK and Akt. SML-10-70-1 inhibits the proliferation of cancer cells H23, H358 and A549 with IC50 of 26.6-47.6 μM .
|
-

- HY-160426
-
|
|
Glycosidase
|
Neurological Disease
|
|
Gcase activator 3 (compound 9Q) is a glucosidase (Glucosidase, GCase) activator that can partially stabilize GCase and increase its activity. Gcase activator 3 reduces mutant GCase protein misfolding and degradation in fibroblasts and dopaminergic midbrain neurons. Gcase activator 3 can be used in the study of Parkinson's disease (PD) and related synucleinopathies .
|
-

- HY-B0248R
-
|
|
Reference Standards
Glucocorticoid Receptor
Huntingtin
Apoptosis
|
Neurological Disease
Inflammation/Immunology
|
|
Desonide (Standard) is the analytical standard of Desonide. This product is intended for research and analytical applications. Desonide is a non-fluorinated corticosteroid anti-inflammatory agent that acts on the glucocorticoid receptor. Desonide can also specifically bind to the mutant huntingtin protein (mHTT), reducing the level and toxicity of mHTT. Desonide can be used in the research of Huntington's disease and inflammatory diseases such as atopic dermatitis.
|
-

- HY-16906
-
|
|
Src
|
Cancer
|
|
T338C Src-IN-2 is a potent mutant c-Src T338C kinase inhibitor with IC50 of 317 nM; also inhibits T338C/V323A and T338C/V323S with IC50 of 57 nM/19 nM.
|
-

- HY-79077R
-
|
|
EGFR
|
Cancer
|
|
Osimertinib (dimesylate) (Standard) is the analytical standard of Osimertinib (dimesylate). This product is intended for research and analytical applications. Osimertinib dimesylate (AZD-9291 dimesylate) is an irreversible and mutant selective EGFR inhibitor with IC50s of 12 and 1 nM against EGFRL858R and EGFRL858R/T790M, respectively.
|
-

- HY-122753
-
|
|
MDM-2/p53
|
Cancer
|
|
SLMP53-1 is a wild-type and mutant p53 reactivator with promising antitumor activity. SLMP53-1 mediates the reprograming of glucose metabolism in cancer cells. SLMP53-1 depletes angiogenesis, decreasing endothelial cell tube formation and vascular endothelial growth factor (VEGF) expression levels .
|
-

- HY-169903
-
|
|
Apoptosis
|
Cancer
|
|
SMIP34 is a PELP1 inhibitor. SMIP34 binds to PELP1 with a Kd of 37.4 μM. SMIP34 inhibits cancer cell proliferation and tumor progression. SMIP34 can be used for breast cancer research, and is active against wild-type (WT), mutant (MT) ER + and therapy-resistant (TR)-ER + breast cancer .
|
-

- HY-168766
-
|
|
Prostaglandin Receptor
COX
|
Inflammation/Immunology
|
|
O-Acetylsalicylhydroxamic acid is an acetylation inhibitor of prostaglandin H2 synthase that can suppress PGE2 synthesis in the body and block the cyclooxygenase activity of PGHS in vitro. O-Acetylsalicylhydroxamic acid requires the presence of the active site residue Ser-529 to act on human PGHS-1; the S529A mutant is resistant to the inactivation effects of this inhibitor .
|
-

- HY-155285
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
YS-363 is a potent, selective, and orally active EGFR inhibitor, with IC50s of 0.96 nM and 0.67 nM for wild-type and L858R mutant forms of EGFR, respectively. YS-363 can induce G0/G1 cell cycle arrest and apoptosis .
|
-

- HY-167642
-
|
(Z)-R278474; (Z)-TMC278; (Z)-DB08864
|
Drug Isomer
HIV
Reverse Transcriptase
|
Infection
|
|
(Z)-Rilpivirine ((Z)-R278474) is the (Z)-isomer of Rilpivirine (HY-10574). Rilpivirine is an effective and selective diarylpyrimidine (DAPY) non-nucleoside reverse transcriptase inhibitor (NNRTI). It exhibits potent antiviral activity against both wild-type HIV (EC50 = 0.4 nM) and mutant strains (EC50 = 0.1-2.0 nM) .
|
-

- HY-145702
-
|
|
MEK
ERK
|
Cancer
|
|
MAP855 is a highly potent, selective, ATP-competitive and orally active MEK1/2 kinase inhibitor (MEK1 ERK2 cascade IC50=3 nM, pERK EC50=5 nM). MAP855 shows equipotent inhibition of wild-type and mutant MEK1/2 .
|
-

- HY-128784
-
|
|
MDM-2/p53
Reactive Oxygen Species (ROS)
|
Cancer
|
|
PK11007 is a mild thiol alkylator with anticancer activity. PK11007 stabilizes p53 via selective alkylation of two surface-exposed cysteines without compromising its DNA binding activity. PK11007 induces mutant p53 cancer cell death by increasing reactive oxygen species (ROS) levels .
|
-

- HY-137971
-
|
|
Ras
|
Cancer
|
|
(±)-Spiro-oxanthromicin A (Compound 4), a polyketide, is a K-Ras inhibitor with an IC50 of 26.7 μM. (±)-Spiro-oxanthromicin A can be isolated from soil-derived Streptomyces sp. (±)-Spiro-oxanthromicin A can mislocalize oncogenic mutant K-Ras from the plasma membrane of intact MDCK cells. (±)-Spiro-oxanthromicin A can be used for cancers research .
|
-

- HY-15772S1
-
|
AZD-9291-13C,d3; Mereletinib-13C,d3
|
EGFR
|
Cancer
|
|
Osimertinib- 13C,d3 is the deuterium and 13C labeled Osimertinib. Osimertinib (AZD9291) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M, respectively.
|
-

- HY-176937
-
|
|
RET
Apoptosis
|
Cancer
|
|
HSN608 is an orally active, potent RET G810 mutant inhibitor. HSN608 inhibits G810C, G810R, G810D. HSN608 induces Apoptosis. HSN608 inhibits the Selpercatinib (HY-114370)-resistant RET(G810C) CDX tumors .
|
-

- HY-123985
-
|
|
Mitochondrial Metabolism
|
Others
|
|
MFN2 agonist-1 (B-A/l) potently stimulates mitochondrial fusion in mitofusin 2 (MFN2)-deficient cells. MFN2 agonist-1 reverses mitochondrial “clumping” (formation of static mitochondrial aggregates) and restores mitochondrial motility in cultured mouse neurons expressing the CMT2A mutant MFN2 T105M.
|
-

- HY-17040
-
|
TMC114; UIC-94017
|
HIV
HIV Protease
|
Infection
Inflammation/Immunology
|
|
Darunavir (TMC114), an orally active next generation HIV protease inhibitor, has a similar antiviral activity against the mutant and the wild-type viruses. Darunavir (TMC114) is potent against laboratory HIV-1 strains and primary clinical isolates (IC50 = 0.003 μM; IC90 = 0.009 μM) with minimal cytotoxicity .
|
-

- HY-147702
-
|
|
Prion Protein
|
Infection
Neurological Disease
|
|
BB 0305179 (compound 59) is a potent anti-prion agent, with an IC50 of 4.7 μM. BB 0305179 inhibits the toxicity of a specific PrP mutant carrying a deletion in the hydrophobic domain . BB 0305179 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-162265
-
|
|
Apoptosis
|
Cancer
|
|
UCM-13369 (Compound 4b) is a NPM1 inhibitor. UCM-13369 downregulates the pathway associated with mutant NPM1 C+, and specifically recognizes the C-end DNA-binding domain of NPM1 C+. UCM-13369 induces apoptosis in AML cell lines and primary cells, that can be used for the research of AML .
|
-

- HY-138301
-
Miclxin
1 Publications Verification
DS37262926
|
Wnt
β-catenin
Apoptosis
|
Cancer
|
|
Miclxin (DS37262926) is a potent inhibitor of mutant β-catenin, involving in Wnt signaling pathway. Miclxin induces β-catenin-dependent apoptosis, leads to severe mitochondrial damage with the loss of mitochondrial membrane. Miclxin kills tumor via targeting to MIC60, a major components of the mitochondrial contact site and cristae organizing system (MICOS) complex .
|
-

- HY-112870
-
|
Alflutinib; Furmonertinib; AST2818
|
EGFR
|
Cancer
|
|
Firmonertinib (Alflutinib; Furmonertinib) is an orally active, mutant-selective, and highly brain penetrant EGFR inhibitor. Firmonertinib inhibits EGFR active mutations as well as the T790M acquired resistant mutation. Firmonertinib has the potential for the research of cancer diseases, especially advanced non-small cell lung cancer (NSCLC) with EGFR ex20ins mutation .
|
-

- HY-134653
-
|
|
Epigenetic Reader Domain
|
Cancer
|
|
5-Ph-IAA is a derivative of IAA. 5-Ph-IAA, a ligand, establishes the auxin-inducible degron 2 (AID2) system together with an OsTIR1 (F74G) mutant. AID2 induces rapid and efficient depletion of mAID-fused proteins to study protein function in living cells, causing tumor suppression .
|
-

- HY-112601
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
IDH1 Inhibitor 1 is a potent, orally bioavailable, brain-penetrant and selective mutant IDH1 inhibitor with IC50s of 0.021 μM, 0.045 μM, and 2.52 μM for IDH1 R132H, IDH1 R132C, and IDH1 WT, respectively . Anticancer activity .
|
-

- HY-15729R
-
|
|
EGFR
|
Cancer
|
|
Rociletinib (Standard) is the analytical standard of Rociletinib. This product is intended for research and analytical applications. Rociletinib (CO-1686) is an orally delivered kinase inhibitor that specifically targets the mutant forms of EGFR including T790M, and the Ki values for EGFRL858R/T790M and EGFRWT are 21.5 nM and 303.3 nM, respectively.
|
-

- HY-15103
-
|
|
PTHR
Androgen Receptor
|
Cancer
|
|
AH3960 (compound 16c) is an antagonist of androgen receptor. AH3960 binds wild as well as T877 mutant type androgen receptors. AH3960 selectively inhibits T877 with an IC50 value of 0.82 μM. AH3960 also serves as an agonist of parathyroid hormone receptor-1 (PTHR1) .
|
-

- HY-119082A
-
|
|
mAChR
|
Others
|
|
VU0029767 is an allosteric enhancer of the M1 muscarinic receptor with the activity to modulate M1 receptor activity. VU0029767 can enhance M1 receptor activity by increasing agonist affinity, but exhibits different properties from other compounds under different experimental conditions, such as effects on mutant M1 receptors and effects on downstream signaling pathways.
|
-

- HY-15772
-
|
AZD-9291; Mereletinib
|
EGFR
|
Cancer
|
|
Osimertinib (AZD9291) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M, respectively. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
|
-

- HY-100028
-
AT-130
2 Publications Verification
|
HBV
DNA/RNA Synthesis
|
Infection
Cancer
|
|
AT-130, a phenylpropenamide derivative, is a potent hepatitis B virus (HBV) replication non-nucleoside inhibitor. AT-130 inhibits the viral DNA synthesis with an EC50 of 0.13 μM. AT-130 inhibits both wt and mutant HBVs. AT-130 has anti-HBV activity in hepatoma cells .
|
-

- HY-12847
-
|
|
Raf
Src
Apoptosis
|
Cancer
|
|
CCT241161 is an orally active pan-RAF inhibitor with IC50s of 3, 6, 10, 15 and 30 nM for LCK, CRAF, SRC, V600E-BRAF and BRAF, respectively. CCT241161 shows good activity to in BRAF and NRAS mutant melanomas. CCT241161 also exhibits anticancer cell proliferative activity .
|
-

- HY-N6790
-
-

- HY-100213
-
EAI045
3 Publications Verification
|
EGFR
|
Cancer
|
|
EAI045 is an allosteric and the fourth-generation inhibitor of mutant EGFR with IC50s of 1.9, 0.019, 0.19 and 0.002 μM for EGFR, EGFR L858R, EGFR T790M and EGFR L858R/T790M at 10 μM ATP, respectively.
|
-

- HY-117187
-
|
|
CFTR
|
Others
|
|
Corr4A is a chemical corrector, which can be used for cystic fibrosis. Corr4A interacts directly with the cystic fibrosis transmembrane conductance regulator (CFTR) or affects indirectly its folding process. Corr4A increases the expression of CFTR ΔF508 on the cell surface, thereby improving its transport to the plasma membrane and increasing the stability of the rescued mutant protein .
|
-

- HY-112870A
-
|
Alflutinib mesylate; Furmonertinib mesylate; AST2818 mesylate
|
EGFR
|
Cancer
|
|
Firmonertinib (Alflutinib; Furmonertinib) mesylate is is an orally active, mutant-selective, and highly brain penetrant EGFR inhibitor. Firmonertinib mesylate inhibits EGFR active mutations as well as the T790M acquired resistant mutation. Firmonertinib mesylate has the potential for the research of cancer diseases, especially advanced non-small cell lung cancer (NSCLC) with EGFR ex20ins mutation .
|
-

- HY-W050122
-
|
|
GABA Receptor
|
Neurological Disease
|
|
S-(+)-GABOB is an endogenous ligand with antiepileptic activity. S-(+)-GABOB is a metabolite of GABA and may function as a neurotransmitter. S-(+)-GABOB behaves as a full agonist when bound to the ρ(1) wild-type receptor. S-(+)-GABOB acts as a competitive antagonist in the ρ(1) T244S mutant receptor .
|
-

- HY-15772A
-
|
AZD-9291 mesylate; Mereletinib mesylate
|
EGFR
|
Cancer
|
|
Osimertinib mesylate (AZD9291 mesylate) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
|
-

- HY-141449
-
|
|
Molecular Glues
β-catenin
|
Cancer
|
|
NRX-103094 is a potent enhancer of the interaction between β-catenin, and its cognate E3 ligase, SCF β-TrCP. NRX-103094 enhances the binding of pSer33/Ser37 β-catenin peptide for β-TrCP with an EC50 of 62 nM and a Kd of 0.6 nM .
|
-

- HY-15244
-
|
BYL-719
|
PI3K
|
Cancer
|
|
Alpelisib (BYL-719) is a potent, selective, and orally active PI3Kα inhibitor. Alpelisib (BYL-719) shows efficacy in targeting PIK3CA-mutated cancer. Alpelisib (BYL-719) also inhibits p110α/p110γ/p110δ/p110β with IC50s of 5/250/290/1200 nM, respectively. Antineoplastic activity .
|
-

- HY-142512
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-24, a potent EGFR inhibitor, shows inhibition against EGFR(del19/T790M/C797S) and EGFR(L858R/T790M/C797S), respectively .
|
-

- HY-137516
-
LC-2
2 Publications Verification
|
PROTACs
Ras
|
Cancer
|
|
LC-2 is a potent and first-in-class von Hippel-Lindau-based PROTAC capable of degrading endogenous KRAS G12C, with DC50s between 0.25 and 0.76 μM . LC-2 covalently binds KRAS G12C with a MRTX849 warhead and recruits the E3 ligase VHL, inducing rapid and sustained KRAS G12C degradation leading to suppression of MAPK signaling in both homozygous and heterozygous KRAS G12C cell lines .
|
-

- HY-10358
-
|
MK-2206 (2HCl)
|
Organoid
Akt
Autophagy
Apoptosis
|
Cancer
|
|
MK-2206 dihydrochloride (MK-2206 (2HCl)) is an orally active, BBB-penetrated allosteric AKT inhibitor with IC50s of 5 nM, 12 nM, and 65 nM for AKT1, AKT2, and AKT3, respectively. MK-2206 dihydrochloride induces autophagy .
|
-

- HY-141448
-
|
|
β-catenin
E1/E2/E3 Enzyme
|
Cancer
|
|
NRX-2663 is an enhancer of the interaction between β-catenin, and its cognate E3 ligase, SCF β-TrCP. NRX-2663 enhances the binding of β-catenin peptide for β-TrCP with an EC50 of 22.9 μM and a Kd of 54.8 nM .
|
-

- HY-152539
-
|
|
HIV
|
Infection
|
|
HIV-1 inhibitor-54 is a potent HIV-1 inhibitor. HIV-1 inhibitor-54 has anti-HIV activity in MT-4 cells against WT HIV-1 (strain IIIB) with an EC50 value of 0.032 μM. HIV-1 inhibitor-54 can be used for the research of virus infection .
|
-

- HY-156712
-
|
|
Antibody-Drug Conjugates (ADCs)
EGFR
|
Cancer
|
|
Depatuxizumab MMAE is an antibody-drug conjugate (ADC) comprising an anti EGFR monoclonal antibody (Depatuxizumab) (HY-P99849) and the cytotoxic agent Monomethyl auristatin E (MMAE) (HY-15162). Depatuxizumab MMAE can be used for the study of EGFR-expressing advanced solid tumors.
|
-

- HY-132842A
-
|
(S)-DZD9008
|
EGFR
Btk
|
Cancer
|
|
(S)-Sunvozertinib ((S)-DZD9008), the S-enantiomer of Sunvozertinib, shows inhibitory activity against EGFR exon 20 NPH and ASV insertions, EGFR L858R/T790M mutation and Her2 exon20 YVMA insertion (IC50=51.2 nM, 51.9 nM, 1 nM, and 21.2 nM, respectively). (S)-Sunvozertinib also inhibits BTK .
|
-

- HY-15244R
-
|
|
PI3K
Reference Standards
|
Cancer
|
|
Alpelisib (Standard) is the analytical standard of Alpelisib. This product is intended for research and analytical applications. Alpelisib (BYL-719) is a potent, selective, and orally active PI3Kα inhibitor. Alpelisib (BYL-719) shows efficacy in targeting PIK3CA-mutated cancer. Alpelisib (BYL-719) also inhibits p110α/p110γ/p110δ/p110β with IC50s of 5/250/290/1200 nM, respectively. Antineoplastic activity .
|
-

- HY-176547
-
|
|
FGFR
Cytochrome P450
|
Endocrinology
|
|
FGFR2/3-IN-3 is a dual-target FGFR2/3 inhibitor with IC50s of 2.7 nM (TEL-FGFR2) and 3.9 nM (TEL-FGFR3), respectively. FGFR2/3-IN-3 has effective activity against both wild-type and mutant FGFR3. FGFR2/3-IN-3 has low CYP3A4 inhibitory effect and hERG toxicity. FGFR2/3-IN-3 improves the imbalance between chondrocyte proliferation and differentiation and promotes bone growth by inhibiting the signaling pathway mediated by mutant FGFR3. FGFR2/3-IN-3 shows a growth-promoting effect in a dwarfism mouse model and has the potential to study bone development disorder-related diseases such as achondroplasia (ACH) .
|
-

- HY-164393
-
|
|
JAK
Bcr-Abl
Apoptosis
|
Cancer
|
|
ON044580, an α-benzoyl styryl benzyl sulfide, is a potent and non-ATP-competitive JAK2 kinase inhibitor with IC50s of 1.23 μM, 1.09 μM for WT and V617F mutant JAK2, respectively. ON044580 inhibits the JAK2 kinase activity either by binding to the STAT-5 binding domain of JAK2 or by binding to an allosteric site. ON044580 exerts its antiproliferative effect in JAK2 V617F-positive leukemic cells. ON044580 effectively induces apoptosis of Imatinib (HY-15463)-resistant chronic myelogenous leukemia (CML) cells. ON044580 also inhibits both WT and T315I mutant forms of the BCR-ABL kinase. ON044580 has the potential for myeloproliferative disorders typified by aberrant JAK/STAT signaling .
|
-

- HY-174311
-
|
|
Glycosidase
|
Neurological Disease
Metabolic Disease
|
|
GT-02216 can bind to GCase allosterically and enhance its activity. GT-02216 enhances the activity of GCase in primary human fibroblasts dose-dependently reduces the accumulation of its substrate, hexosylsphingosine (HexCer). GT-02216 reduces the Tau accumulation in mutant GBA1 fibroblasts. GT-02216 can be used for the study of Parkinson’s disease .
|
-

- HY-162265A
-
|
|
Apoptosis
|
Cancer
|
|
UCM-13369 (Compound 4b) is a NPM1 inhibitor. UCM-13369 downregulates the pathway associated with mutant NPM1 C+, and specifically recognizes the C-end DNA-binding domain of NPM1 C+. UCM-13369 induces apoptosis in AML cell lines and primary cells. UCM-13369 can be used for leukemia research .
|
-

- HY-112301
-
|
BLU-667
|
RET
|
Cancer
|
|
Pralsetinib (BLU-667) is a highly potent, selective RET inhibitor. Pralsetinib (BLU-667) inhibits WT RET, RET mutants V804L, V804M, M918T and CCDC6-RET fusion with IC50s of 0.4, 0.3, 0.4, 0.4, and 0.4 nM, respectively .
|
-

- HY-149318
-
|
|
Gap Junction Protein
Connexin
|
Others
|
|
VRT-534 (Compound 3) is a chemical chaperone targeting connexin 26 (Cx26). Vrt-534 exhibits dose-responsive binding to recombinant WT Cx26 and mutant Cx26 K188N with EC50s of 19 and 5 μM, respectively. Vrt-534 can be used in research related to hearing impairment .
|
-

- HY-139150
-
|
|
Histone Methyltransferase
|
Cancer
|
|
EZH2-IN-4 is an orally active, potent EZH2 inhibitor with IC50s of 0.923 nM and 2.65 nM against wild type (WT) 5-membered (5-mer) EZH2 and mutant 5-mer EZH2, respectively. EZH2-IN-4 has anti-cancer activity .
|
-

- HY-147942
-
|
|
PROTACs
EGFR
|
Cancer
|
|
MS9449 is a potent PROTAC EGFR degrader with Kds of 17 nM and 10 nM for EGFR WT and EGFR L858R, respectively. MS9449 effectively induces degradation of mutant EGFRs through both the ubiquitin/proteasome system (UPS) and autophagy/lysosome pathways. MS9449 potently inhibits the proliferation of NSCLC cells. MS9449 can be used for researching anticancer .
|
-

- HY-149607
-
|
|
SHP2
|
Cancer
|
|
SHP2-IN-22 is SHP2 allosteric inhibitor with an IC50 value of 17.7 nM. SHP2-IN-22 inhibits the proliferation, migration, and invasion of MIA PaCa-2 pancreatic cancer cells. SHP2-IN-22 can be used for Kirsten rat sarcoma viral oncogene (KRAS) mutant cancer research .
|
-

- HY-121845
-
|
|
HSP
|
Metabolic Disease
|
|
4-Br-Bnlm is a selective inhibitor of glucose-regulated protein 94 (Grp94) with an EC50 value of 0.96 µM. 4-Br-Bnlm reduces the levels of mutant myocilin proteins as well as wild-type myocilin misfold in cells. 4-Br-Bnlm promotes the clearance of toxic formsof myocilin and reduces myocilin toxicity .
|
-

- HY-E70827
-
|
|
FGFR
|
Cancer
|
|
FGFR2 is a receptor tyrosine kinase that is involved in many human cancers such as intrahepatic cholangiocarcinomas and hepatocellular carcinomas. FGFR2 V564L is a mutant of FGFR3. FGFR2 V564L Recombinant Human Active Protein Kinase is a recombinant FGFR2 V564L protein that can be used to study FGFR2 V564L-related functions .
|
-

- HY-13912
-
|
|
Wnt
Porcupine
Casein Kinase
|
Cancer
|
|
IWP-2 is an inhibitor of Wnt processing and secretion with an IC50 of 27 nM. IWP-2 targets the membrane-bound O-acyltransferase porcupine (Porcn) and thus preventing a crucial Wnt ligand palmitoylation. IWP-2 is also an ATP-competitive CK1δ inhibitor with an IC50 of 40 nM for the gatekeeper mutant M82FCK1δ .
|
-

- HY-15801
-
|
|
Anaplastic lymphoma kinase (ALK)
EGFR
|
Cancer
|
|
HG-14-10-04 is a potent ALK and mutant EGFR inhibitor with IC50s of 20 nM, 15.6 nM, 22.6 nM and 124.5 nM for ALK, EGFR LR/TM, EGFR 19del/TM/CS and EGFR LR/TM/CS, respectively. HG-14-10-04 can be used to research anticancer .
|
-

- HY-18690S
-
|
AG-221-d6
|
Isotope-Labeled Compounds
Isocitrate Dehydrogenase (IDH)
|
Others
|
|
Enasidenib-d6 (AG-221-d6) is the deuterium labeled Enasidenib (HY-18690) . Enasidenib is an oral, potent, reversible, selective inhibitor of the IDH2 mutant enzymes, with IC50s of 100 and 400 nM against IDH2 R140Q and IDH2 R172K, respectively .
|
-

- HY-155537
-
|
YK-029A
|
EGFR
|
Cancer
|
|
YK-029A is an orally active inhibitor of mutant EGFR,targeting to both the T790M mutations (EGFR T790M) and exon 20 insertion of EGFR (EGFRex20ins). YK-029A exhibits significant antitumor activity,and results tumor regression in EGFRex20ins-driven PDX models .
|
-

- HY-163110
-
|
|
HIV
Reverse Transcriptase
|
Infection
|
|
NNRT-IN-2 (compound 7w) is an orally available non-nucleoside reverse transcriptase inhibitor (NNRTI) with broad inhibitory effects on wild-type HIV-1 and mutant strains. NNRT-IN-2 inhibits HIV-1 reverse transcriptase with an EC50 of 22 nM. NNRT-IN-2 is insensitive to CYP and hERG and has good safety and pharmacokinetic characteristics .
|
-

- HY-177273
-
|
|
Bcr-Abl
|
Cancer
|
|
Kinase modulator-1 (P142, MS: m/z445) is a kinase modulator. Kinase modulator-1 can inhibit Bcr-Abl activity, especially against drug-resistant mutant kinases (such as T315I Bcr-Abl). Kinase modulator-1 can be used for cancers like chronic myelogenous leukemia (CML) research .
|
-

- HY-14829A
-
|
D-Isofagomine hydrochloride; Isofagomine hydrochloride
|
Glycosidase
|
Metabolic Disease
|
|
Afegostat hydrochloride (D-Isofagomine hydrochloride) is a potent β-galactosidase inhibitor with activity ameliorating GM1-gangliosidosis and Morquio B disease-associated mutations. Afegostat hydrochloride is able to induce the maturation of mutant β-galactosidase in fibroblasts from patients with GM1-gangliosidosis. Afegostat hydrochloride also promotes the reduction of keratin sulfate and oligosaccharide load in patient cells .
|
-

- HY-128306
-
|
|
HCV Protease
DNA/RNA Synthesis
|
Infection
|
|
HCV-IN-50 (Compound 2) is a competitive and selective HCV NS5B RNA-dependent RNA polymerase inhibitor with an IC50 of 0.3 μM for NS5B △C21 enzyme over △C55 enzyme. HCV-IN-50 has an antiviral activity and efficiently blocks replication of HCV subgenomic replicons especially mutant replicons .
|
-

- HY-128350A
-
|
|
Farnesyl Transferase
γ-Glutamyl Transferase (GGT)
|
Cancer
|
|
FGTI-2734 mesylate is a RAS C-terminal mimetic dual farnesyl transferase (FT) and geranylgeranyl transferase-1 (GGT) inhibitor with IC50s of 250 nM and 520 nM for FT and GGT, respectively. FGTI-2734 mesylate can prevent membrane localization of KRAS, hence solving KRAS resistance problem and thwarting mutant KRAS patient-derived pancreatic tumors .
|
-

- HY-15292
-
S107
1 Publications Verification
|
Calcium Channel
|
Cardiovascular Disease
|
|
S107 is an orally available, blood brain barrier-permeable compound, which stabilizes RyR2 channels by enhancing the binding of calstabin 2 to the mutant Ryr2-R2474S channel. S107 inhibits Ca 2+ leakage from the sarcoplasmic reticulum (SR) and prevents cardiac arrhythmias and raises the seizure threshold .
|
-

- HY-144053
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-36 is a potent EGFR inhibitor with IC50s of 19.09 nM, 120.01 nM, 2.35 nM for EGFR (WT), HER2 (WT), HER2 (A775_G776insYVMA), respectively. EGFR-IN-36 has potential for wild and/or mutant EGFR and/or HER2 kinase mediated tumors research .
|
-

- HY-147941
-
|
|
PROTACs
EGFR
|
Cancer
|
|
MS9427 is a potent PROTAC EGFR degrader with Kds of 7.1 nM and 4.3 nM for EGFR WT and EGFR L858R, respectively. MS9427 selectively degrades the mutant but not the WT EGFR through both the ubiquitin/proteasome system (UPS) and autophagy/lysosome pathways. MS9427 potently inhibits the proliferation of NSCLC cells. MS9427 can be used for researching anticancer .
|
-

- HY-130608
-
|
|
EGFR
|
Cancer
|
|
Mutated EGFR-IN-3 (compound 3) is a potent, ATP-competitive and highly selective allosteric dibenzodiazepinone inhibitor of the EGFR(L858R/T790M) and EGFR(L858R/T790M/C797S) mutants with IC50 values of 12 nM and 13 nM, respectively .
|
-

- HY-175743
-
|
|
FGFR
|
Cancer
|
|
TYRA-200 is an orally active FGFR1/2/3 inhibitor. TYRA-200 shows dose-dependent tumor regression in both wild-type FGFR2 and FGFR2 mutant mice model. TYRA-200 can be used for advanced or metastatic intrahepatic cholangiocarcinoma and other FGFR2-driven solid tumors research .
|
-

- HY-E70647
-
|
|
Anaplastic lymphoma kinase (ALK)
|
Cancer
|
|
Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase in the insulin receptor superfamily. ALK has multiple mutants. ALK R1275Q is commonly found in neuroblastoma (NB). ALK R1275Q Recombinant Human Active Protein Kinase is a recombinant ALK R1275Q protein that can be used to study ALK R1275Q-related functions .
|
-

- HY-18161
-
|
|
FLT3
Aurora Kinase
|
Cancer
|
|
CCT241736 is a potent and orally bioavailable dual FLT3 and Aurora kinase inhibitor, which inhibits Aurora kinases (Aurora-A Kd, 7.5 nM, IC50, 38 nM; Aurora-B Kd, 48 nM), FLT3 kinase (Kd, 6.2 nM), and FLT3 mutants including FLT3-ITD (Kd, 38 nM) and FLT3(D835Y) (Kd, 14 nM).
|
-

- HY-117273
-
|
|
Raf
Autophagy
|
Cancer
|
|
AZ304 is an ATP-competitive dual BRAF kinase inhibitor, potently inhibits wild type BRAF, V600E mutant BRAF and wild type CRAF, with IC50s of 79 nM, 38 nM and 68 nM, respectively. AZ304 also has significant effect on other kinases, such as p38 (IC50, 6 nM), CSF1R (IC50, 35 nM). Anti-tumor activity .
|
-

- HY-125147
-
|
|
NAMPT
|
Cancer
|
|
A-1293201 is a substrate-independent NAMPT inhibitor with antitumor activity. A-1293201 effectively reduces the total cellular NAD +/NADH (NADt) level, subsequently leading to ATP depletion and cancer cell death. In addition, A-1293201 can effectively overcome the acquired resistance mechanism of the NAMPT Y18 mutant to CHS-828 (HY-10079) .
|
-

- HY-15772S
-
|
AZD-9291-d6; Mereletinib-d6
|
EGFR
|
Cancer
|
|
Osimertinib-d6 is a deuterium labeled osimertinib. Osimertinib is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
|
-

- HY-158991
-
|
|
CFTR
|
Inflammation/Immunology
|
|
I1421 is an activator of the cystic fibrosis transmembrane conductance regulator (CFTR) with an EC50 of 64 nM for WT CFTR currents. I1421 also allosterically activates multiple mutants causing cystic fibrosis (CF) with good in vivo potency, with an oral bioavailability of 60% in mice corresponding to a half-life of 75 min. I1421 synergizes with Elexacaftor (HY-111772) to enhance CFTR currents .
|
-

- HY-E70649
-
|
|
Raf
|
Cancer
|
|
BRAF is a member of the Raf kinase family of growth signal transduction protein kinases. BRAF has multiple mutants. BRAF V600E is commonly found in melanoma. BRAF V600E Recombinant Human Active Protein Kinase is a recombinant BRAF V600E protein that can be used to study BRAF V600E-related functions .
|
-

- HY-172780
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
EGFR-IN-161 (Compound DD-8) is a potent and reversible inhibitor of L858R/T790M/C797S mutant EGFR kinases, with an IC50 of 0.87 nM. EGFR-IN-161 can induce apoptosis process, G1-phase arrestation, and migration inhibition in tumor cells .
|
-

- HY-153386
-
|
|
Anaplastic lymphoma kinase (ALK)
|
Cancer
|
|
CPD-1224 is an orally effective ALK inhibitor, a derivative of an ALK inhibitor that connects to the cereblon ligand. CPD-1224 targets the EML4-ALK oncogenic fusion protein and degrades both ALK and the mutant forms L1196M/G1202R. CPD-1224 can slow down tumor growth .
|
-

- HY-10206
-
|
MP470; HPK 56
|
c-Kit
PDGFR
RAD51
FLT3
c-Met/HGFR
RET
Apoptosis
|
Cancer
|
|
Amuvatinib (MP470) is an orally bioavailable multi-targeted tyrosine kinase inhibitor with potent activity against mutant c-Kit, PDGFRα, Flt3, c-Met and c-Ret. Amuvatinib (MP470) is also a DNA repair suppressor through suppression of DNA repair protein RAD51, thereby disrupting DNA damage repair . Antineoplastic activity .
|
-

- HY-142452
-
|
|
Raf
|
Cancer
|
|
Pan-RAF kinase inhibitor 1 is a potent inhibitor of Pan-RAF kinase. Pan-RAF kinase inhibitor 1 regulates MAPK signaling by inhibiting RAF kinase, thereby exerting an effect on the proliferation of RAS-mutant tumor cells. Pan-RAF kinase inhibitor 1 has the potential for the research of cancer diseases (extracted from patent WO2021110141A1, compound 16B) .
|
-

- HY-E70746
-
|
|
MEK
|
Cancer
|
|
MEK1 is the MAP/ERK kinase. The MEK1 kinase directly phosphorylates ERK2, after the activation loop of MEK1 is itself phosphorylated by Raf. MEK1 SESE is a mutant of MEK1. MEK1 SESE Recombinant Human Active Protein Kinase is a recombinant MEK1 SESE protein that can be used to study MEK1 SESE-related functions .
|
-

- HY-10206A
-
|
MP470 hydrochloride; HPK 56 hydrochloride
|
c-Kit
PDGFR
RAD51
FLT3
c-Met/HGFR
RET
|
Cancer
|
|
Amuvatinib hydrochloride (MP470 hydrochloride) is an orally bioavailable multi-targeted tyrosine kinase inhibitor with potent activity against mutant c-Kit, PDGFRα, Flt3, c-Met and c-Ret. Amuvatinib hydrochloride (MP470 hydrochloride) is also a DNA repair suppressor through suppression of DNA repair protein RAD51, thereby disrupting DNA damage repair . Antineoplastic activity .
|
-

- HY-12026
-
|
|
EGFR
|
Cancer
|
|
WZ4002 is a mutant selective EGFR inhibitor with IC50s of 2, 8, 3 and 2 nM for EGFR L858R, EGFR L858R/T790M, EGFR E746_A750 and EGFR E746_A750/T790M, respectively.
|
-

- HY-15729AR
-
|
|
EGFR
|
Cancer
|
|
Rociletinib hydrobromide (Standard) is the analytical standard of Rociletinib hydrobromide. This product is intended for research and analytical applications. Rociletinib hydrobromide (CO-1686 hydrobromide) is an orally delivered kinase inhibitor that specifically targets the mutant forms of EGFR including T790M, and the Ki values for EGFRL858R/T790M and EGFRWT are 21.5 nM and 303.3 nM, respectively.
|
-

- HY-178042
-
|
|
Ras
|
Cancer
|
|
SS-3091 is a pan KRas inhibitor. SS-3091 binds to the interaction interfaces of KRas, destabilizing the ARaf/KRas complex and thereby influencing downstream signaling transduction. SS-3091 has significant anticancer and antiproliferation activities against the KRas G12D, G12C, G12V, and G12S mutants in various cancer cells .
|
-

- HY-E70785
-
|
|
Trk Receptor
|
Cancer
|
|
TRKA is a member of the tropomyosin receptor kinase (TRK) family, and its primary binding ligand is nerve growth factor (NGF). TRKA G667C is a mutant of TRKA. TRKA G667C Recombinant Human Active Protein Kinase is a recombinant TRKA G667C protein that can be used to study TRKA G667C-related functions .
|
-

- HY-153663
-
|
|
Ras
|
Cancer
|
|
TH-Z827 is a mutant selective KRAS(G12D) inhibitor with an IC50 of 2.4 μM. TH-Z827 does not bind KRAS(WT) or KRAS(G12C). TH-Z827 blocked the KRAS(G12D)-CRAF interaction with an IC50 value of 42 μM .
|
-

- HY-161633
-
|
|
PROTACs
EGFR
FAK
|
Cancer
|
|
PROTAC EGFR degrader 11 (Compound B71) is a PROTAC degrader for epidermal growth factor receptor (EGFR), with DC50 <100 nM. PROTAC EGFR degrader 11 binds CRBN-DDB1 with a Ki of 36 nM. PROTAC EGFR degrader 11 degrades EGFR, focal adhesion kinase (FAK), and RSK1, inhibits the proliferation of BaF3 wild type and EGFR mutants, with IC50 <100 nM.
|
-

- HY-178057
-
|
|
EGFR
Akt
Anaplastic lymphoma kinase (ALK)
Apoptosis
|
Cancer
|
|
EGFR-IN-176 is an orally active and ATP-competitive EGFR mutant inhibitor (particularly C797S-mediated EGFR triple mutant). EGFR-IN-176 effectively inhibits subsequent AKT signaling and induces apoptosis in Ba/F3 and PC-9 cells expressing EGFR 19del/T790M/C797S and EGFR L858R/T790M/C797S. EGFR-IN-176 selectively inhibits EGFR signaling in cell lines harboring EGFR triple mutation and shows no inhibitory effect against A431 cells that express wild-type EGFR. EGFR-IN-176 can effectively inhibit the enzymatic activity of ALK (IC50 < 0.5 nM). EGFR-IN-176 can be used for the study of non-small cell lung cancer (NSCLC) .
|
-

- HY-146078
-
|
|
Bacterial
|
Infection
|
|
Antimicrobial agent-1 (compound 6C) possesses potent activity against TolC mutant E. coli with an MIC value of 2 μg/mL. Antimicrobial agent-1 and Colistin exhibit synergistic activity against Gram-negative bacteria. Antimicrobial agent-1 has no cytotoxicity on mammalian cell lines, with MICs > 128 μg/mL in Caco-2 and Vero cell lines .
|
-

- HY-174826
-
|
|
EGFR
c-Kit
|
Cancer
|
|
EGFR-IN-164 (Compound 4) is a selective and covalent allosteric EGFR inhibitor. EGFR-IN-164 significantly inhibits the activity of EGFR L858R/T790M/C797S kinase (IC50: 48.1 nM) and proliferation of of EGFR-mutant cells. EGFR-IN-164 can be used for drug resistance of cancer research .
|
-

- HY-E70818
-
|
|
FGFR
|
Cancer
|
|
FGFR2 is a receptor tyrosine kinase that is involved in many human cancers such as intrahepatic cholangiocarcinomas and hepatocellular carcinomas. FGFR2 K659M is a mutant of FGFR3. FGFR2 K659M Recombinant Human Active Protein Kinase is a recombinant FGFR2 K659M protein that can be used to study FGFR2 K659M-related functions .
|
-

- HY-E70823
-
|
|
FGFR
|
Cancer
|
|
FGFR2 is a receptor tyrosine kinase that is involved in many human cancers such as intrahepatic cholangiocarcinomas and hepatocellular carcinomas. FGFR2 N549K is a mutant of FGFR3. FGFR2 N549K Recombinant Human Active Protein Kinase is a recombinant FGFR4 N535K protein that can be used to study FGFR4 N535K-related functions .
|
-

- HY-161895
-
|
|
CFTR
|
Others
|
|
CFTR corrector 14 (Compound SVQ26) is a class 3 corrector for cystic fibrosis transmembrane conductance regulator (CFTR), that promotes the CFTR activity (EC50 of 3.08 μM with presence of C1 class corrector VX-809). CFTR corrector 14 regulates the mutant-caused misfolding and impaired function of the CFTR protein. CFTR corrector 14 can be used in research about cystic fibrosis .
|
-

- HY-12001
-
|
|
EGFR
|
Cancer
|
|
WZ3146 is a mutant selective EGFR inhibitor with IC50s of
2, 2, 5, 14 and 66 nM for EGFR L858R, EGFR L858R/T790M, EGFR E746_A750, EGFR E746_A750/T790M and EGFR, respectively.
|
-

- HY-128350
-
|
|
Farnesyl Transferase
|
Cancer
|
|
FGTI-2734 is a RAS C-terminal mimetic dual farnesyl transferase (FT) and geranylgeranyl transferase-1 (GGT-1) inhibitor with IC50s of 250 nM and 520 nM for FT and GGT-1, respectively. FGTI-2734 can prevent membrane localization of KRAS, hence solving KRAS resistance problem and thwarting mutant KRAS patient-derived pancreatic tumors .
|
-

- HY-E70646
-
|
|
Anaplastic lymphoma kinase (ALK)
|
Cancer
|
|
Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase in the insulin receptor superfamily. ALK can be used for the study of non-small cell lung carcinoma (NSCLC). ALK has multiple mutants. ALK L1196M Recombinant Human Active Protein Kinase is a recombinant ALK L1196M protein that can be used to study ALK L1196M-related functions .
|
-

- HY-119082
-
|
|
mAChR
|
Others
|
|
(E/Z)-VU0029767 is an allosteric enhancer of M1 muscarinic receptors with the activity to modulate M1 receptor activity. (E/Z)-VU0029767 can enhance M1 receptor activity by increasing agonist affinity, but exhibits different properties from other compounds under different experimental conditions, such as effects on mutant M1 receptors and effects on downstream signaling pathways.
|
-

- HY-116804
-
|
|
Histone Methyltransferase
|
Cancer
|
|
ZLD1039 is a potent, highly selective, and orally bioavailable EZH2 inhibitor. ZLD1039 shows potent and concentration-dependent inhibition of PRC2 enzymatic activity against EZH2 wild-type as well as Y641F, and A677G mutant enzymes with IC50 values of 5.6, 15, and 4.0 nM, respectively. ZLD1039 inhibits breast tumor growth and metastasis .
|
-

- HY-174417
-
|
|
Reverse Transcriptase
HIV
Potassium Channel
Cytochrome P450
|
Infection
|
|
NNRT-IN-10 is a potent, selective and orally active non-nucleoside HIV-1 reverse transcriptase (NNRTI) inhibitor with with an EC50 values ranging from 1.16 to 18.3 nM for HIV and its mutant strains. NNRT-IN-10 exhibits good pharmacokinetic properties and favorable safety profiles. NNRT-IN-10 can be used for the study of AIDS, caused by HIV-1 .
|
-

- HY-E70643
-
|
|
Anaplastic lymphoma kinase (ALK)
|
Cancer
|
|
Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase in the insulin receptor superfamily. ALK has multiple mutants. ALK-F1174L is commonly found in neuroblastoma (NB). ALK-F1174L Recombinant Human Active Protein Kinase is a recombinant ALK-F1174L protein that can be used to study ALK-F1174L-related functions .
|
-

- HY-128862
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-7 is a potent, selective and orally active EGFR kinase inhibitor. EGFR-IN-7 has inhibitory effect for for EGFR (WT) and EGFR (mutant C797S/T790M/L858R) with IC50 values of 7.92 nM and 0.218 nM, respectively. EGFR-IN-7 can be used for the research of various cancers .
|
-

- HY-E70815
-
|
|
FGFR
|
Cancer
|
|
FGFR2 is a receptor tyrosine kinase that is involved in many human cancers such as intrahepatic cholangiocarcinomas and hepatocellular carcinomas. FGFR2 K526E is a mutant of FGFR3. FGFR2 K526E Recombinant Human Active Protein Kinase is a recombinant FGFR2 K526E protein that can be used to study FGFR2 K526E-related functions .
|
-

- HY-19834
-
|
GDC-0853
|
Btk
|
Cancer
|
|
Fenebrutinib (GDC-0853) is a potent, selective, orally available, and noncovalent bruton's tyrosine kinase (Btk) inhibitor with Kis of 0.91 nM, 1.6, 1.3, 12.6, and 3.4 nM for WT Btk, and the C481S, C481R, T474I, T474M mutants. Fenebrutinib has the potential for rheumatoid arthritis and systemic lupus erythematosus research .
|
-

- HY-P10438
-
|
|
Raf
|
Cancer
|
|
TAT-Braftide is a peptide inhibitor designed to block the dimerization of BRAF, thereby inhibiting its kinase activity. The destruction of BRAF dimer by TAT-Braftide makes BRAF protein more susceptible to proteasome degradation, directly inhibits the activity of BRAF kinase, and reduces the activation of MAPK signaling pathway. Tat-braftide can be used for the role of RAF kinase in MAPK signaling pathway and for the study of BRAF mutant cancers .
|
-

- HY-161632
-
|
|
PROTACs
EGFR
FAK
|
Cancer
|
|
PROTAC EGFR degrader 10 (Compound B56) is a PROTAC degrader for epidermal growth factor receptor (EGFR), with DC50 <100 nM. PROTAC EGFR degrader 10 binds CRBN-DDB1 with a Ki of 37 nM. PROTAC EGFR degrader 10 degrades EGFR, focal adhesion kinase (FAK), and RSK1, inhibits the proliferation of BaF3 wild type and EGFR mutants, with IC50 <150 nM .
|
-

- HY-E70644
-
|
|
Anaplastic lymphoma kinase (ALK)
|
Cancer
|
|
Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase in the insulin receptor superfamily. ALK can be used for the study of non-small cell lung carcinoma (NSCLC). ALK has multiple mutants. ALK F1174S Recombinant Human Active Protein Kinase is a recombinant ALK F1174S protein that can be used to study ALK F1174S-related functions .
|
-

- HY-E70825
-
|
|
FGFR
|
Cancer
|
|
FGFR2 is a receptor tyrosine kinase that is involved in many human cancers such as intrahepatic cholangiocarcinomas and hepatocellular carcinomas. FGFR2 V564F is a mutant of FGFR3. FGFR2 V564F Recombinant Human Active Protein Kinase is a recombinant FGFR2 V564F protein that can be used to study FGFR2 V564F-related functions .
|
-

- HY-139995
-
|
|
Drug Intermediate
|
Others
|
|
Spermine precursor-1 (Compound 1) is a redox-sensitive spermine precursor for the potential research of snyder robinson syndrome. Spermine precursor-1 inhibits wild-type (CMS-24949) and spermine synthase gene (SMS) mutant (CMS-26559, and CMS-6233) fibroblast cells with IC50s of 326.7, 198.5, and 244.1 μM, respectively .
|
-

- HY-E70826
-
|
|
FGFR
|
Cancer
|
|
FGFR2 is a receptor tyrosine kinase that is involved in many human cancers such as intrahepatic cholangiocarcinomas and hepatocellular carcinomas. FGFR2 V564I is a mutant of FGFR3. FGFR2 V564I Recombinant Human Active Protein Kinase is a recombinant FGFR2 V564I protein that can be used to study FGFR2 V564I-related functions .
|
-

- HY-173329
-
|
|
Ras
|
Cancer
|
|
KRAS-IN-41 is an inhibitor of KRAS with IC50 values of <0.01 μM for KRAS G12D and KRAS G12V. KRAS-IN-41 inhibits RAS mutant cell lines, GP2D (KRAS-G12D) and SW620 (KRAS-G12V). KRAS-IN-41 can be used in cancer research .
|
-

- HY-E70814
-
|
|
FGFR
|
Cancer
|
|
FGFR2 is a receptor tyrosine kinase that is involved in many human cancers such as intrahepatic cholangiocarcinomas and hepatocellular carcinomas. FGFR2 C491S is a mutant of FGFR3. FGFR2 C491S Recombinant Human Active Protein Kinase is a recombinant FGFR2 C491S protein that can be used to study FGFR2 C491S-related functions .
|
-

- HY-E70642
-
|
|
Anaplastic lymphoma kinase (ALK)
|
Cancer
|
|
Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase in the insulin receptor superfamily. ALK can be used for the study of non-small cell lung carcinoma (NSCLC). ALK has multiple mutants. ALK C1156Y Recombinant Human Active Protein Kinase is a recombinant ALK C1156Y protein that can be used to study ALK C1156Y-related functions .
|
-

- HY-E70817
-
|
|
FGFR
|
Cancer
|
|
FGFR2 is a receptor tyrosine kinase that is involved in many human cancers such as intrahepatic cholangiocarcinomas and hepatocellular carcinomas. FGFR2 K641R is a mutant of FGFR3. FGFR2 K641R Recombinant Human Active Protein Kinase is a recombinant FGFR2 K641R protein that can be used to study FGFR2 K641R-related functions .
|
-

- HY-158409
-
|
|
Molecular Glues
Ras
Cyclophilin
|
Cancer
|
|
Pan-rasin-2 (compound 6A) is a molecular glues that targets RAS. Pan-rasin-2 has significant inhibitory activity on cell proliferation of RAS mutant cell lines. Pan-rasin-2 can form ternary complexes with cyclophilin A (CYPA) and RAS (ON) proteins and the formation of ternary complexes can block the binding of RAF downstream of RAS, which has anti-tumor effects .
|
-

- HY-14174
-
|
|
γ-secretase
|
Neurological Disease
Cancer
|
|
MRK-560, a chemical probe, is an orally active, brain barrier-penetrated γ-Secretase inhibitor, reducing Aβ peptide in rat brain and cerebrospinal fluid. MRK-560 decreases mutant NOTCH1 processing by selectively inhibiting PSEN1. MRK-560 can be used in studies of Alzheimer's disease and T-cell acute lymphoblastic leukaemia (T-ALL) .
|
-

- HY-N0328R
-
|
α-Mangostin (Standard)
|
Fungal
Reactive Oxygen Species (ROS)
Bacterial
Apoptosis
Virus Protease
Reference Standards
|
Cancer
|
|
alpha-Mangostin (Standard) is the analytical standard of alpha-Mangostin. This product is intended for research and analytical applications. alpha-Mangostin (α-Mangostin) is a dietary xanthone with broad biological activities, such as antioxidant, anti-allergic, antiviral, antibacterial, anti-inflammatory and anticancer effects. It is an inhibitor of mutant IDH1 (IDH1-R132H) with a Ki of 2.85 μM.
|
-

- HY-E70822
-
|
|
FGFR
|
Cancer
|
|
FGFR2 is a receptor tyrosine kinase that is involved in many human cancers such as intrahepatic cholangiocarcinomas and hepatocellular carcinomas. FGFR2 N549H is a mutant of FGFR3. FGFR2 N549H Recombinant Human Active Protein Kinase is a recombinant FGFR4 N535H protein that can be used to study FGFR4 N535H-related functions .
|
-

- HY-19985A
-
|
|
EGFR
|
Cancer
|
|
(3S, 4S)-PF-06459988 is the S enantiomer of PF-06459988 with less active. PF-06459988 is a potent irreversible inhibitor of T790M mutant epidermal growth factor receptor (EGFR). PF-06459988 has excellent selectivity against EGFR wild-type while possessing a minimally reactive electrophile that reduces the propensity of off-target labeling .
|
-

- HY-W653989
-
|
|
Isotope-Labeled Compounds
|
Others
|
|
Desonide- 13C3 is the 13C labeled isotope of Desonide (HY-B0248). Desonide is a non-fluorinated corticosteroid anti-inflammatory agent that acts on the glucocorticoid receptor. Desonide can also specifically bind to the mutant huntingtin protein (mHTT), reducing the level and toxicity of mHTT. Desonide can be used in the research of Huntington's disease and inflammatory diseases such as atopic dermatitis .
|
-

- HY-147841
-
|
|
HIV
Reverse Transcriptase
|
Infection
|
|
HIV-1 inhibitor-41 (Compound B23) is an orally active non-nucleoside HIV-1 reverse transcriptase inhibitor with EC50 values of 20.8 nM and 50 nM against HIV-1 WT and mutant E138K strain, respectively. HIV-1 inhibitor-41 shows low hERG, no apparent CYP enzymatic inhibition and no acute toxicity .
|
-

- HY-114358
-
|
ONO-7475
|
TAM Receptor
Trk Receptor
|
Cancer
|
|
Tamnorzatinib (ONO-7475) is a potent, selective, and orally active Axl/Mer inhibitor with IC50 values of 0.7 nM and 1.0 nM, respectively. Tamnorzatinib sensitizes AXL-overexpressing EGFR-mutant NSCLC cells to the EGFR-TKIs, suppresses the emergence and maintenance of tolerant cells. Tamnorzatinib combines with Osimertinib (HY-15772) provides a bright promise for the study of EGFR-mutated non-small cell lung cancer (NSCLC).
|
-

- HY-E70645
-
|
|
Anaplastic lymphoma kinase (ALK)
|
Cancer
|
|
Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase in the insulin receptor superfamily. ALK can be used for the study of non-small cell lung carcinoma (NSCLC). ALK has multiple mutants. ALK G1202R Recombinant Human Active Protein Kinase is a recombinant ALK G1202R protein that can be used to study ALK G1202R-related functions .
|
-

- HY-E70820
-
|
|
FGFR
|
Cancer
|
|
FGFR2 is a receptor tyrosine kinase that is involved in many human cancers such as intrahepatic cholangiocarcinomas and hepatocellular carcinomas. FGFR2 L617V is a mutant of FGFR3. FGFR2 L617V Recombinant Human Active Protein Kinase is a recombinant FGFR2 L617V protein that can be used to study FGFR2 L617V-related functions .
|
-

- HY-E70821
-
|
|
FGFR
|
Cancer
|
|
FGFR2 is a receptor tyrosine kinase that is involved in many human cancers such as intrahepatic cholangiocarcinomas and hepatocellular carcinomas. FGFR2 M537I is a mutant of FGFR3. FGFR2 M537I Recombinant Human Active Protein Kinase is a recombinant FGFR2 M537I protein that can be used to study FGFR2 M537I-related functions .
|
-

- HY-E70824
-
|
|
FGFR
|
Cancer
|
|
FGFR2 is a receptor tyrosine kinase that is involved in many human cancers such as intrahepatic cholangiocarcinomas and hepatocellular carcinomas. FGFR2 R612T is a mutant of FGFR3. FGFR2 R612T Recombinant Human Active Protein Kinase is a recombinant FGFR2 R612T protein that can be used to study FGFR2 R612T-related functions .
|
-

- HY-E70819
-
|
|
FGFR
|
Cancer
|
|
FGFR2 is a receptor tyrosine kinase that is involved in many human cancers such as intrahepatic cholangiocarcinomas and hepatocellular carcinomas. FGFR2 K659N is a mutant of FGFR3. FGFR2 K659N Recombinant Human Active Protein Kinase is a recombinant FGFR2 K659N protein that can be used to study FGFR2 K659N-related functions .
|
-

- HY-152160
-
|
|
HIV
Reverse Transcriptase
|
Infection
|
|
HIV-1 invistor-50 is a non-nucleoside reverse transcriptase inhibitor (NNRTI) that targets HIV-1 reverse transcriptase (RT) (IC50=50 nM). HIV-1 inhibitor-50 shows significant antiviral activity, with EC50s of 2.22-53.3nM against HIV-1 IIIB and its mutant strains .
|
-

- HY-E70809
-
|
|
Btk
|
Inflammation/Immunology
Cancer
|
|
Bruton's tyrosine kinase (BTK) plays a vital role in B-cell antigen receptor (BCR) signalling transduction pathway. BTK can be used for the study of lymphomas and autoimmune diseases. BTK has multiple mutants. BTK V416L Recombinant Human Active Protein Kinase is a recombinant BTK V416L protein that can be used to study BTK V416L-related functions .
|
-

- HY-E70816
-
|
|
FGFR
|
Cancer
|
|
FGFR2 is a receptor tyrosine kinase that is involved in many human cancers such as intrahepatic cholangiocarcinomas and hepatocellular carcinomas. FGFR2 K641N is a mutant of FGFR3. FGFR2 K641N Recombinant Human Active Protein Kinase is a recombinant FGFR2 K641N protein that can be used to study FGFR2 K641N-related functions .
|
-

- HY-101119
-
|
|
Estrogen Receptor/ERR
|
Cancer
|
|
GLL398 is a potent, orally bioavailable selective estrogen receptor degrader (SERD) with an IC50 value of 1.14 nM. GLL398 shows a strong dose-dependent binding to ER with a mutation at Y537S (IC50=29.5 nM). GLL398 blocks tumor growth in xenograft models of breast cancer.
|
-

- HY-178211
-
|
|
Ras
MEK
ERK
Akt
|
Cancer
|
|
SHY-867 is a pan RAS inhibitor. SHY-867 effectively prevents the binding of K-Ras proteins and other members of the Ras superfamily of small GTPases with EC50 values of 0.5-3 μM. SHY-867 effectively inhibits the phosphorylation of MEK, ERK1/2, and AKT downstream of K-Ras. SHY-867 inhibits the formation of the Ras-GTP activity complex. SHY-867 can be used to the studies of pancreatic cancer and non-small cell lung cancer .
|
-

- HY-178194
-
|
|
Ras
MEK
ERK
Akt
|
Cancer
|
|
SHY-855 is a pan RAS inhibitor. SHY-855 effectively prevents the binding of K-Ras proteins and other members of the Ras superfamily of small GTPases with IC50 values of 0.3-5 μM. SHY-855 effectively inhibits the phosphorylation of MEK, ERK1/2, and AKT downstream of K-Ras. SHY-855 inhibits the formation of the Ras-GTP activity complex. SHY-855 can be used to the studies of pancreatic cancer and non-small cell lung cancer .
|
-

- HY-114900
-
|
|
Antibiotic
|
Infection
Others
|
|
BB-3497 is a potent, orally active and selective peptide deformylase (PDF) inhibitor. BB-3497 is highly selective for PDF (IC50 = 7 nM for E. coli PDF.Ni) over the other mammalian metalloenzymes (MMP-1/2/3/7 and enkephalinase). BB-3497 exhibits potent activity against gram-positive bacteria and some gram-negative pathogens. BB-3497 protects mice from infection in systemic models of Staphylococeus aureus. BB-3497 can be used for anti-bacterial infection research .
|
-

- HY-P11356
-
|
|
IFNAR
|
Cancer
|
|
KRAS G12V Peptide is a specific peptide derived from the Kirsten rat sarcoma virus (KRAS) gene carrying the G12V oncogenic mutation. KRAS G12V Peptide induces responses like IFN-γ secretion and cytotoxicity. KRAS G12V Peptide can be used for the study of immune responses against KRAS G12V-mutant tumors .
|
-

- HY-10683
-
|
|
PI3K
mTOR
|
Cancer
|
|
PKI-402 is a selective, reversible, ATP-competitive inhibitor of PI3K, including PI3K-α mutants, and mTOR (IC50=2, 3, 7,14 and 16 nM for PI3Kα, mTOR, PI3Kβ, PI3Kδ and PI3Kγ).
|
-

- HY-145574
-
|
WX-0593
|
Anaplastic lymphoma kinase (ALK)
ROS Kinase
BCRP
Akt
ERK
STAT
|
Cancer
|
|
Iruplinalkib (WX-0593) is an orally active and selective ALK/ROS1 inhibitor. Iruplinalkib can effectively inhibit tyrosine autophosphorylation of ALK and mutant ALK, EGFR, with the IC50 between 5.38 and 16.74 nM. Iruplinalkib is also a suppressive agent of the transporter MATE1, MATE2K, P-gp and BCRP. Iruplinalkib can be used in the study of non-small cell lung cancer .
|
-

- HY-174293
-
|
|
Drug Derivative
|
Cancer
|
|
Osimertinib dimer is a dimer of Osimertinib (HY-15772). Osimertinib (AZD9291) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M, respectively. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
|
-

- HY-160023
-
|
D3S-001
|
Ras
|
Cancer
|
|
Elisrasib is an orally active inhibitor for KRAS. Elisrasib inhibits the proliferation of KRAS G12C mutant H358 and MIA-PA-CA-2 with an IC50 of 0.6 and 0.44 nM. Elisrasib exhibits good metabolic stability in hepatocytes, liver microsomes, plasma and whole blood in various species. D3S-001 exhibits good pharmacokinetic characteristics and antitumor efficacy in mice .
|
-

- HY-10574S
-
|
|
Isotope-Labeled Compounds
HIV
Reverse Transcriptase
|
Infection
|
|
Rilpivirine-d6 is the deuterium labeled Rilpivirine. Rilpivirine (R278474) is a potent and specific diarylpyrimidine (DAPY) non-nucleoside reverse transcriptase inhibitor (NNRTI). Rilpivirine has high antiviral activity against wild-type HIV (EC50=0.4 nM) and mutant viruses (EC50=0.1-2.0 nM). Rilpivirine has a high genetic barrier to resistance development of HIV .
|
-

- HY-E70720
-
|
|
FGFR
|
Cancer
|
|
FGFR3 activating mutations are drivers of malignancy in several human tissues, including bladder, lung, cervix, and blood. FGFR3 K650M is a mutant of FGFR3. FGFR3 K650M Recombinant Human Active Protein Kinase is a recombinant FGFR3 K650M protein that can be used to study FGFR3 K650M-related functions .
|
-

- HY-177454
-
|
|
ADC Linker
|
Others
|
|
Basivarsen linker is a linker used in HY-177452 Zeleciment basivarsen for coupling a TfR1-binding Fab (HY-P990780 Zeleciment) and an antisense oligonucleotide. Zeleciment basivarsen is an antibody-oligonucleotide conjugate (AOC) designed to target mutant nuclear myotonic dystrophy protein kinase (DMPK) RNA for RHase H-mediated degradation to correct splicing. It is used for the study of myotonic dystrophy type 1 (DM1).
|
-

- HY-111517
-
|
|
Apoptosis
SF3B1
|
Cancer
|
|
H3B-8800 is a potent and orally active SF3B splicing modulator. H3B-8800 direct interaction with the SF3b complex and shows anti-cancer activity. H3B-8800 has the potential for the research of acute myeloid leukemia (AML) with SF3B1 mutant .
|
-

- HY-130259
-
|
|
Atg8/LC3
Autophagy
|
Neurological Disease
|
|
LC3-mHTT-IN-AN2 (Compound AN2) is a mHTT-LC3 linker compound, which interacts with both mutant huntingtin protein (mHTT) and LC3B but not with wtHTT or irrelevant control proteins. LC3-mHTT-IN-AN2 reduces the levels of mHTT in an allele-selective manner in cultured Huntington disease (HD) mouse neurons .
|
-

- HY-147941A
-
|
|
PROTACs
EGFR
|
Cancer
|
|
MS9427 TFA is a potent PROTAC EGFR degrader with Kds of 7.1 nM and 4.3 nM for EGFR WT and EGFR L858R, respectively. MS9427 TFA selectively degrades the mutant but not the WT EGFR through both the ubiquitin/proteasome system (UPS) and autophagy/lysosome pathways. MS9427 TFA potently inhibits the proliferation of NSCLC cells. MS9427 TFA can be used for researching anticancer .
|
-

- HY-130426
-
|
Mal-PEG3-acid
|
ADC Linker
PROTAC Linkers
|
Cancer
|
|
Maleimido-tri(ethylene glycol)-propionic acid is a cleavable ADC linker used in the synthesis of antibody-drug conjugates (ADCs). Maleimido-tri(ethylene glycol)-propionic acid is used for the preparation of neolymphostin-based ADC precursors for site-specific cysteine mutant trastuzumab-A114C conjugation . Maleimido-tri(ethylene glycol)-propionic acid also can be used as a PEG-based PROTAC linker that can be used in the synthesis of PROTACs.
|
-

- HY-146112
-
|
|
IRAK
|
Cancer
|
|
IRAK4-IN-14 (compound 28) is a potent, selective and orally active IRAK4 inhibitor with an IC50 of 0.003 µM. IRAK4-IN-14 shows good PK parameters in rats and mouse. IRAK4-IN-14 shows synergistic in vitro activity against MyD88/CD79 double mutant ABC-DLBCL in combination with Acalabrutinib .
|
-

- HY-179039
-
|
|
EGFR
|
Cancer
|
EGFR-IN-183 (Compound 5q) is an EGFR inhibitor. EGFR-IN-183 strongly binds to EGFR (T790M/L858R) mutants. EGFR-IN-183 exhibits anti-proliferative activity against MDA-MB-231, with an IC50 value of 16.4 μM. EGFR-IN-183 can be used for research on triple-negative breast cancer .
|
-

- HY-P11357
-
|
|
IFNAR
|
Cancer
|
|
KRAS G12C Peptide is a specific peptide derived from the Kirsten rat sarcoma virus (KRAS) gene carrying the G12C oncogenic mutation. KRAS G12C Peptide induces responses like IFN-γ secretion and cytotoxicity. KRAS G12C Peptide can be used for the study of immune responses against KRAS G12C-mutant tumors .
|
-

- HY-174274
-
|
|
ERK
|
Cancer
|
|
ERK2-IN-6 (Compound 20) is a highly selective ERK1/2 inhibitor with an IC50 value of 7.9 nM for ERK2. ERK2-IN-6 inhibits the proliferation of BRAF V600E mutant cells (IC50=250 nM in A375 cells). ERK2-IN-6 is promising for research of solid tumors, such as BRAF-mutated melanoma .
|
-

- HY-E70808
-
|
|
Btk
|
Inflammation/Immunology
Cancer
|
|
Bruton's tyrosine kinase (BTK) plays a vital role in B-cell antigen receptor (BCR) signalling transduction pathway. BTK can be used for the study of lymphomas and autoimmune diseases. BTK has multiple mutants. BTK C481R Recombinant Human Active Protein Kinase is a recombinant BTK C481R protein that can be used to study BTK C481R-related functions .
|
-

- HY-130258
-
|
|
Atg8/LC3
Autophagy
|
Neurological Disease
|
|
LC3-mHTT-IN-AN1 (Compound AN1) is a mHTT-LC3 linker compound, which interacts with both mutant huntingtin protein (mHTT) and LC3B but not with wtHTT or irrelevant control proteins. LC3-mHTT-IN-AN1 reduces the levels of mHTT in an allele-selective manner in cultured Huntington disease (HD) mouse neurons .
|
-

- HY-174216
-
|
|
DNA/RNA Synthesis
Oxidative Phosphorylation
Mitochondrial Metabolism
|
Neurological Disease
Metabolic Disease
|
|
PZL-A is a activator of mitochondrial DNA (mtDNA) synthesis. PZL-A restores wild-type-like activity to mutant forms of polymerase γ (POLγ) with AC50 s of 160 and 20 nM for A467T and G848S. PZL-A activates mtDNA synthesis in cells, enhancing biogenesis of the oxidative phosphorylation machinery and cellular respiration. PZL-A is promising for relieving POLG disease and other severe conditions linked to depletion of mtDNA .
|
-

- HY-178448
-
|
|
EGFR
JAK
Ferroptosis
Apoptosis
Reactive Oxygen Species (ROS)
Cannabinoid Receptor
Glutathione Peroxidase
Caspase
|
Inflammation/Immunology
Cancer
|
|
EGFR-IN-178 is an orally active EGFR mutant inhibitor, exhibits highly selective inhibitory activity against mutants of the EGFR enzyme, including Del19 (IC50 = 3.4 nM), L858R/T790 M (IC50 = 2.9 nM), and Del19/T790 M (IC50 = 2.5 nM). EGFR-IN-178 has good activity against JAK2 (IC50 = 55.6 nM) and JAK3 (IC50 = 46.1 nM) kinases. EGFR-IN-178 can increase cellular lipid oxide MDA, meanwhile decrease GSH content, causing ferroptosis in cancer cells. EGFR-IN-178 promotes apoptosis by increasing cleaved caspase-3 expression. EGFR-IN-178 can inhibit the phosphorylation of EGFR protein and decrease the active form p-JAK2 for JAK2, induce an increase in intracellular ROS. EGFR-IN-178 can be used for the study of non-small cell lung cancer (NSCLC) .
|
-

- HY-168916
-
|
|
SARS-CoV
Virus Protease
|
|
|
Jun13296 is an orally active quinoline SARS-CoV-2 papain-like protease inhibitor (IC50 = 0.13 µM, Ki = 8.8 nM). Jun13296 exhibits potent inhibition against SARS-CoV-2 variants and Nirmatrelvir (HY-138687)-resistant mutants. Jun13296 improves lung viral titers, and prevents lung tissue damage in a SARS-CoV-2 infection model .
|
-

- HY-E70763
-
|
|
PDGFR
|
Cancer
|
|
PDGFRalpha is a receptor tyrosine kinases that stimulate cell survival, proliferation and motility. Upon PDGF binding, the receptor dimerizes and undergoes a conformational change, which activates the kinase domain. PDGFRalpha T674I is a mutant of PDGFRalpha. PDGFRalpha T674I Recombinant Human Active Protein Kinase is a recombinant PDGFRalpha T674I protein that can be used to study PDGFRalpha T674I-related functions .
|
-

- HY-15772AR
-
|
AZD-9291 mesylate (Standard); Mereletinib mesylate (Standard)
|
Reference Standards
EGFR
|
Cancer
|
|
Osimertinib (mesylate) (Standard) is the analytical standard of Osimertinib (mesylate). This product is intended for research and analytical applications. Osimertinib mesylate (AZD9291 mesylate) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
|
-

- HY-174131
-
|
|
Estrogen Receptor/ERR
|
Cancer
|
|
PVTX-321 (Compound 16a) is an orally active estrogen receptor α (ERα) degrader. PVTX-321 can potently degrade ERα (DC50=0.15 nM in MCF-7 cells) and also has inhibitory activity against mutant ERα (IC50=59 nM). PVTX-321 is promising for research of ER+/HER2- breast cancer .
|
-

- HY-15164A
-
|
BPI-2009
|
EGFR
|
Cancer
|
|
Icotinib (BPI-2009) is a potent and specific EGFR inhibitor with an IC50 of 5 nM; also inhibits mutant EGFR L858R, EGFR L858R/T790M, EGFR T790M and EGFR L861Q. Icotinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-178763
-
|
|
JAK
|
Inflammation/Immunology
|
|
DEL1187-126-28-16 is a JAK3 inhibitor with IC50s of 10 and 0.97 nM against JAK3 WT in 0 and 60 min. DEL1187-126-28-16 has no activity at all towards the C909S mutant, JAK1, JAK2, and TYK2. DEL1187-126-28-16 can be used for the study of immune-mediated diseases .
|
-

- HY-169388
-
|
|
AUTACs
|
Cancer
|
|
YT 6-2 is the p62/SQSTM1 targeting, autophagy-targeting ligand (ATL) of AUTOTAC ATC-324 (HY-169385), which is an bivalent AR (Androgen Receptor) degrader. ATC-324 can reduce nuclear AR levels and downregulate target gene expression of AR and AR-v7, and also has a degradation effect on common AR mutants in PCa .
|
-

- HY-E70762
-
|
|
PDGFR
|
Cancer
|
|
PDGFRalpha is a receptor tyrosine kinases that stimulate cell survival, proliferation and motility. Upon PDGF binding, the receptor dimerizes and undergoes a conformational change, which activates the kinase domain. PDGFRalpha D842V is a mutant of PDGFRalpha. PDGFRalpha D842V Recombinant Human Active Protein Kinase is a recombinant PDGFRalpha D842V protein that can be used to study PDGFRalpha D842V-related functions .
|
-

- HY-15772R
-
|
AZD-9291 (Standard); Mereletinib (Standard)
|
Reference Standards
EGFR
|
Cancer
|
|
Osimertinib (Standard) is the analytical standard of Osimertinib. This product is intended for research and analytical applications. Osimertinib (AZD9291) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M, respectively. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
|
-

- HY-E70744
-
|
|
MEK
|
Cancer
|
|
MEK1 is the MAP/ERK kinase. The MEK1 kinase directly phosphorylates ERK2, after the activation loop of MEK1 is itself phosphorylated by Raf. MEK1 F53L is a mutant of MEK1. MEK1 F53L Recombinant Human Active Protein Kinase is a recombinant MEK1 F53L protein that can be used to study MEK1 F53L-related functions .
|
-

- HY-75368
-
|
|
LRRK2
|
Neurological Disease
|
|
SRI-31255 is an orally active LRRK2 inhibitor with IC50 values of 520 and 427 nM for human wild-type (WT) and mutant G2019S, respectively. SRI-31255 exerts neuroprotective effects by binding to the ATP-binding pocket of LRRK2, inhibiting kinase activity. SRI-31255 can be used as a lead compound for the development of LRRK2-targeted drugs for the treatment of Parkinson's disease .
|
-

- HY-N12109
-
|
|
Others
|
Others
|
|
Albizziin is an amino acid analog with activity as a competitive inhibitor of asparagine synthetase. Albizziin has been used to isolate Chinese hamster ovary cell mutants with altered levels of the target enzyme. Several mutational classes of albizziin can be distinguished based on cross-resistance to β-aspartic hydroxamic acid. Studies on asparagine synthetase have shown that resistance to albizziin may be associated with altered regulation of asparagine synthetase, structural mutations of the enzyme, and gene amplification .
|
-

- HY-E70786
-
|
|
Akt
|
Cancer
|
|
AKT is the oncogenic protein kinase that regulates essential cellular functions such as migration, proliferation, differentiation, apoptosis, and metabolism. Mammalian cells are characterized by the expression of three different Akt isoforms, Akt1, Akt2, and Akt3. AKT1 E17K is a mutant of AKT1. AKT1 E17K Recombinant Human Active Protein Kinase is obtained by expressing AKT1 E17K proteins .
|
-

- HY-168069
-
|
|
Parasite
Dihydrofolate reductase (DHFR)
|
Infection
|
|
DHFR-IN-20 (Compound LA1) is a Plasmodium falciparum dihydrofolate reductase (DHFR) inhibitor, with Kis of 0.16, 0.30, 6.6 nM for PfDHFR-WT, PfDHFR-QM, HsDHFR. DHFR-IN-20 has antimalarial activities (IC50: 1.4 nM and 1.6 μM for P. falciparum carrying the wild-type (TM4/8.2) and the quadruple mutant (V1/S) PfDHFR enzyme .
|
-

- HY-15164
-
|
BPI-2009H
|
EGFR
|
Cancer
|
|
Icotinib Hydrochloride (BPI-2009) is a potent and specific EGFR inhibitor with an IC50 of 5 nM; also inhibits mutant EGFR L858R, EGFR L858R/T790M, EGFR T790M and EGFR L861Q. Icotinib (Hydrochloride) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-17040R
-
|
|
HIV
HIV Protease
|
Infection
Inflammation/Immunology
|
|
Darunavir (Standard) is the analytical standard of Darunavir. This product is intended for research and analytical applications. Darunavir (TMC114), an orally active next generation HIV protease inhibitor, has a similar antiviral activity against the mutant and the wild-type viruses. Darunavir (TMC114) is potent against laboratory HIV-1 strains and primary clinical isolates (IC50 = 0.003 μM; IC90 = 0.009 μM) with minimal cytotoxicity .
|
-

- HY-164487
-
|
|
Proteasome
|
Cancer
|
|
JBJ-08-178-01 is a mutant-selective tyrosine kinase inhibitor against human epidermal growth factor receptor 2 (HER2) with an antitumoral activity. JBJ-08-178-01 reduces both the kinase activity and protein levels of HER2 by inducing proteasomal degradation of the receptor in lung cancer. JBJ-08-178-01 is promising for research of non-small-cell lung cancer .
|
-

- HY-178755
-
|
|
JAK
|
Inflammation/Immunology
|
|
DEL1187-126-5-80 is a JAK3 inhibitor with IC50s of 52 and 7.9 nM against JAK3 WT in 0 and 60 min. DEL1187-126-5-80 has no activity at all towards the C909S mutant, JAK1, JAK2, and TYK2. DEL1187-126-5-80 can be used for the study of immune-mediated diseases .
|
-

- HY-146113
-
|
|
IRAK
|
Cancer
|
|
IRAK4-IN-15 (compound 35) is a potent and selective IRAK4 inhibitor with an IC50 of 0.002 µM. IRAK4-IN-15 shows good human PK predictions with low intrinsic clearance. IRAK4-IN-15 shows great synergistic in vitro activity against MyD88/CD79 double mutant ABC-DLBCL in combination with Acalabrutinib. .
|
-

- HY-175870
-
|
|
Ras
|
Cancer
|
|
pan-KRAS-IN-18 (Compound 14-1) is a pan-KRAS inhibitor. pan-KRAS-IN-18 has potent antitumor activities and significantly inhibits the proliferation of wild-type and mutant (such as KRAS G12D, KRAS G12V and KRAS G12C) cancer cells. pan-KRAS-IN-18 effectively inhibits tumor growth in GP2D and Panc0403 xenograft mouse models .
|
-

- HY-179102
-
|
|
SARS-CoV
Virus Protease
|
Infection
|
|
SARS-CoV-2 Mpro-IN-51 is a SARS-CoV-2 Mpro inhibitor (IC50 = 26 nM). SARS-CoV-2 Mpro-IN-51 has inhibitory effects on the triple mutant L50F/E166A/L167F. SARS-CoV-2 Mpro-IN-51 can be used for research on viral infections .
|
-

- HY-123418
-
|
|
Phosphoglycerate Dehydrogenase (PHGDH)
|
Cancer
|
|
PKUMDL-WQ-2201 is a PHGDH non-NAD+-competing allosteric inhibitor (IC50=35.7 μM). PKUMDL-WQ-2201 also inhibits PHGDH mutants with IC50s of 69 μM (T59A) and >300 μM (T56AK57A), respectively. PKUMDL-WQ-2201 inhibits de novo serine synthesis in cancer cells, and reduces tumor growth .
|
-

- HY-E70764
-
|
|
PDGFR
|
Cancer
|
|
PDGFRalpha is a receptor tyrosine kinases that stimulate cell survival, proliferation and motility. Upon PDGF binding, the receptor dimerizes and undergoes a conformational change, which activates the kinase domain. PDGFRalpha V561D is a mutant of PDGFRalpha. PDGFRalpha V561D Recombinant Human Active Protein Kinase is a recombinant PDGFRalpha V561D protein that can be used to study PDGFRalpha V561D-related functions .
|
-

- HY-146746
-
|
|
HIV
|
Infection
|
|
HIV-1 inhibitor-19 is a potent HIV-1 non-nucleoside reverse transcriptase inhibitor (NNRTI). HIV-1 inhibitor-19 maintains its inhibitory activity against L100I, K103N and V106A/ F227L mutant strains with EC50s of 7.3 nM, 9.2 nM and 21.0 nM, respectively.
|
-

- HY-E70745
-
|
|
MEK
|
Cancer
|
|
MEK1 is the MAP/ERK kinase. The MEK1 kinase directly phosphorylates ERK2, after the activation loop of MEK1 is itself phosphorylated by Raf. MEK1 P124L is a mutant of MEK1. MEK1 P124L Recombinant Human Active Protein Kinase is a recombinant MEK1 P124L protein that can be used to study MEK1 P124L-related functions .
|
-

- HY-139590
-
|
BOS-172738; DS-5010
|
RET
PDGFR
|
Cancer
|
|
Zeteletinib (BOS-172738; DS-5010) is an orally active, selective RET kinase inhibitor with nanomolar potency against RET and >300-fold selectivity against VEGFR2. Zeteletinib shows exquisite potency for the wild type RET, RET V804M/L gatekeeper mutants, and the most common oncogenic RET mutation M918T. Zeteletinib has potent antitumor activity .
|
-

- HY-P1708
-
|
BRN 537924; NSC 657143
|
Bacterial
|
Infection
|
|
Enopeptin A, originally isolated from a culture broth of Streptomyces sp. RK-1051, is a depsipeptide antibiotic that contains two unusual amino acids (N-methylalanine and 4-methylproline) and features a pentaenone side chain. It is effective against Gram-positive bacteria, including methicillin-resistant S. aureus (MIC=25 μg/mL), and Gram-negative bacteria, including mutant forms of E. coli and P. aeruginosa (MICs=200 μg/mL); however, it is not inhibitory to fungi.
|
-

- HY-10574S1
-
|
R278474-13C6; TMC278-13C6; DB08864-13C6
|
Isotope-Labeled Compounds
HIV
Reverse Transcriptase
|
Infection
|
|
Rilpivirine- 13C6 (R278474- 13C6) is 13C labeled Rilpivirine. Rilpivirine (R278474) is a potent and specific diarylpyrimidine (DAPY) non-nucleoside reverse transcriptase inhibitor (NNRTI). Rilpivirine has high antiviral activity against wild-type HIV (EC50=0.4 nM) and mutant viruses (EC50=0.1-2.0 nM). Rilpivirine has a high genetic barrier to resistance development of HIV .
|
-

- HY-174254
-
|
|
Akt
Apoptosis
Caspase
PARP
β-catenin
|
Cancer
|
|
AKT-IN-28 is an Akt allosteric inhibitor, a derivative of Shikonin (HY-N0822). AKT-IN-28 effectively binds to the allosteric site of Akt through hydrophobic and hydrogen interactions with Kd of 2.07 μM. AKT-IN-28 significantly inhibits Akt activity, induces cell apoptosis, arrests cell cycle in G2/M phase, and suppresses proliferation, migration and metabolism of KRAS mutant colorectal cancer cells .
|
-

- HY-402410
-
TETi76
1 Publications Verification
|
TET Protein
|
Cancer
|
|
TETi76 is an orally active TET family inhibitor with IC50 values ??of 1.5, 9.4 and 8.8 μM for TET1, TET2 and TET3, respectively. TETi76 competitively binds to the active site of TET enzymes, reduces cytosine hydroxymethylation and restricts clonal growth of TET2 mutants in vitro and in vivo, but does not affect the growth of normal hematopoietic precursor cells. TETi76 can be used for leukemia research .
|
-

- HY-155227
-
|
|
Anaplastic lymphoma kinase (ALK)
EGFR
Apoptosis
|
Cancer
|
|
ALK/EGFR-IN-1 (Compound 8l) is an ALK/EGFR dual inhibitor that blocks the phosphorylation of EGFR and ALK. ALK/EGFR-IN-1 inhibits ALK/EGFR mutants respectively, with IC50 of 4.3 nM for EGFR L858R T790M in H1975 cells and EML4-ALK in BaF3 cells, respectively. and 3.6 nM. ALK/EGFR-IN-1 may be used in NSCLC research .
|
-

- HY-176523
-
|
|
Ras
|
Cancer
|
|
KRAS G12D inhibitor 29 (Compound Formula (I)) is an orally active and selective KRAS G12D mutant inhibitor. KRAS G12D inhibitor 29 blocks downstream signaling pathways mediated by KRAS G12D, suppressing tumor cell proliferation. KRAS G12D inhibitor 29 is promising for research of KRAS G12D mutation-related cancers (such as pancreatic cancer, lung cancer, colorectal cancer) .
|
-

- HY-10206R
-
|
MP470 (Standard); HPK 56 (Standard)
|
Reference Standards
c-Kit
PDGFR
RAD51
FLT3
c-Met/HGFR
RET
Apoptosis
|
Cancer
|
|
Amuvatinib (Standard) is the analytical standard of Amuvatinib. This product is intended for research and analytical applications. Amuvatinib (MP470) is an orally bioavailable multi-targeted tyrosine kinase inhibitor with potent activity against mutant c-Kit, PDGFRα, Flt3, c-Met and c-Ret. Amuvatinib (MP470) is also a DNA repair suppressor through suppression of DNA repair protein RAD51, thereby disrupting DNA damage repair . Antineoplastic activity .
|
-

- HY-E70776
-
|
|
RET
|
Cancer
|
|
Rearranged during transfection (RET) is a transmembrane receptor protein tyrosine kinase (PTK) activated normally by forming ternary complexes with its cognate ligands and co-receptors. RET alterations by point mutations and gene fusions were found in diverse cancers. RET S891A is a mutant of RET. RET S891A Recombinant Human Active Protein Kinase is a recombinant RET S891A protein that can be used to study RET S891A-related functions .
|
-

- HY-139590A
-
|
BOS-172738 hemiadipate; DS-5010 hemiadipate
|
RET
PDGFR
|
Cancer
|
|
Zeteletinib (BOS-172738; DS-5010) hemiadipate is an orally active, selective RET kinase inhibitor with nanomolar potency against RET and >300-fold selectivity against VEGFR2. Zeteletinib hemiadipate shows exquisite potency for the wild type RET, RET V804M/L gatekeeper mutants, and the most common oncogenic RET mutation M918T. Zeteletinib hemiadipate has potent antitumor activity .
|
-

- HY-158325
-
|
|
PROTACs
FLT3
Apoptosis
|
Cancer
|
|
PROTAC FLT-3 degrader 4 is an orally active CRBN-based FLT3-PROTAC degrader that potently induces FLT3-ITD degradation through the ubiquitin-proteasome system. PROTAC FLT-3 degrader 4 shows highly selective to FLT3-ITD mutant acute myeloid leukemia (AML) cells. (Blue: CRBN ligand, Black: linker; Pink: FLT3 inhibitor) .
|
-

- HY-164492
-
|
|
Raf
|
Cancer
|
|
LSN3074753, an analog of LY3009120 (HY-12558), is a pan-RAF and Raf dimer inhibitor. LSN3074753 demonstrates activity against tumor cells with MAPK pathway activation driven by BRAF monomer or RAF dimers including BRAF- or KRAS-mutant colorectal cancer. LSN3074753 combined with Cetuximab (HY-P9905) shows additive and synergistic effects for colorectal cancer PDX models, particularly those with KRAS or BRAF mutation .
|
-

- HY-124089S
-
|
|
Isotope-Labeled Compounds
Cannabinoid Receptor
|
Metabolic Disease
|
|
Eicosapentaenoyl ethanolamide-d4 is the deuterium labeled Eicosapentaenoyl ethanolamide. Eicosapentaenoyl ethanolamide, an omega-3 fatty acid, is one of N-acylethanolamines (NAEs). Eicosapentaenoyl ethanolamide is cannabinoid CB1/CB2 receptor agonist. Eicosapentaenoyl ethanolamide acts as a metabolic signal. Eicosapentaenoyl ethanolamide inhibits dietary restriction (DR)-induced lifespan extension in wild type animals and suppresses lifespan extension in a TOR pathway mutant .
|
-

- HY-18343
-
|
|
MDM-2/p53
Apoptosis
|
Cancer
|
|
CP-31398 can stabilize the active conformation of p53 and promote p53 activity in cancer cells with either mutant or wild-type p53. In addition, CP-31398 can upregulate p53 target genes, such as p21WAF1/Cip1 and KILLER/DR5. CP-31398 exerts an inhibitory effect on tumor growth .
|
-

- HY-149318B
-
|
|
Connexin
Gap Junction Protein
|
Others
|
|
(R,R)-VRT-534 is the (R,R)-enantiomer of VRT-534 (HY-149318). VRT-534 (Compound 3) is a chemical chaperone targeting connexin 26 (Cx26). Vrt-534 exhibits dose-responsive binding to recombinant WT Cx26 and mutant Cx26 K188N with EC50s of 19 and 5 μM, respectively. Vrt-534 can be used in research related to hearing impairment .
|
-

- HY-156020
-
|
|
Glycosidase
|
Metabolic Disease
|
|
Glucocerebrosidase-IN-2 (compound 12) is a quinazoline analogue and an inhibitor of glucocerebrosidase (GC). Glucocerebrosidase-IN-2 has the potential to improve GC translocation to lysosomes in Gaucher disease patient-derived cells (mostly carrying the N370S mutation). Glucocerebrosidase-IN-2 inhibits the hydrolysis of 4-methylumbelliferone β-D-glucopyranoside (4MU) and fluorescent glycosylceramide (FlourGC) in N370S mutant tissues with an AC50 of 25.29 μM .
|
-

- HY-16767S1
-
|
MK-1439-13C,d3
|
Isotope-Labeled Compounds
HIV
Reverse Transcriptase
|
Infection
|
|
Doravirine-13C,d3 (MK-1439-13C,d3) is the deuterium labeled Doravirine (HY-16767). Doravirine (MK-1439) is a highly specific HIV-1 nonnucleoside reverse transcriptase inhibitor with IC50s of 4.5 nM, 5.5 nM and 6.1 nM against the wild type and K103N and Y181C reverse transcriptase mutants, respectively .
|
-

- HY-149555
-
|
|
Eukaryotic Initiation Factor (eIF)
|
Neurological Disease
|
|
DNL343 is a potent, selective, orally active and brain-penetrant activator of eukaryotic initiation factor eIF2B. DNL343 inhibits the activity of the integrated stress response (ISR) in the central nervous system (CNS) and reverses neurodegeneration and neuroinflammation. DNL343 also prevents motor dysfunction and premature death in eIF2B loss-of-function (LOF) mutant mice. DNL343 can be used in the study of neurodegenerative diseases .
|
-

- HY-143218
-
|
Tetraphenylethene maleimide
|
Huntingtin
Parasite
|
Infection
Neurological Disease
|
|
TPE-MI (Tetraphenylethene maleimide) is a thiol probe for measuring unfolded protein load and proteostasis in cells (the excitation wavelength is 350 nm and the emission wavelength is 470 nm). TPE-MI can report imbalances in proteostasis in induced pluripotent stem cell models of Huntington disease, as well as cells transfected with mutant Huntington exon 1 before the formation of visible aggregates. TPE-MI also detects protein damage following dihydroartemisinin research of the malaria parasitesPlasmodium falciparum .
|
-

- HY-17598
-
|
|
Oxidative Phosphorylation
Parasite
p38 MAPK
Raf
Apoptosis
|
Infection
Cancer
|
|
Rafoxanide is a poent, orally active halogenated salicylaniline agent with antiparasitic activity. Rafoxanide interferes with energy metabolism in trematodes by uncoupling oxidative phosphorylation. Rafoxanide is also found to be a potent inhibitor of the BRAF V600E mutant protein, which is important in colorectal cancer. Rafoxanide can be used for the control of infestation with Hemonchus species or Fasciola species in sheep and cattle as well as Oestrus ovis in sheep. Rafoxanide can also be used for cancer research .
|
-

- HY-112585
-
|
TMC114-d9; UIC-94017-d9
|
HIV
HIV Protease
|
Infection
Inflammation/Immunology
|
|
Darunavir-d9 (TMC114-d9) is the deuterium labeled Darunavir. Darunavir (TMC114), an orally active next generation HIV protease inhibitor, has a similar antiviral activity against the mutant and the wild-type viruses. Darunavir (TMC114) is potent against laboratory HIV-1 strains and primary clinical isolates (IC50 = 0.003 μM; IC90 = 0.009 μM) with minimal cytotoxicity .
|
-

- HY-101663
-
|
MK-8408
|
HCV
|
Infection
|
|
Ruzasvir (MK-8408) is an orally active inhibitor for genotypic HCV nonstructural protein 5A (NS5A), which inhibits the replication of NS5A mutants GT1a, GT1a L31V, GT1a Y93H, GT2b, GT3a and GT4a with EC90 ranging from 0.003 to 0.067 nM. Ruzasvir exhibits good pharmacokinetic characters in rhesus monkey model .
|
-

- HY-E70778
-
|
|
RET
|
Cancer
|
|
Rearranged during transfection (RET) is a transmembrane receptor protein tyrosine kinase (PTK) activated normally by forming ternary complexes with its cognate ligands and co-receptors. RET alterations by point mutations and gene fusions were found in diverse cancers. RET V804L is a mutant of RET. RET V804L Recombinant Human Active Protein Kinase is a recombinant RET V804L protein that can be used to study RET V804L-related functions .
|
-

- HY-107779
-
|
|
Raf
|
Cancer
|
|
BI-882370 is a potent and selective RAF kinase inhibitor that binds to the ATP binding site of the kinase positioned in the DFG-out (inactive) conformation of the BRAF kinase. BI-882370 (BI 882370) inhibits the oncogenic BRAF V600E-mutant, the WT BRAF and CRAF kinases with IC50s of 0.4, 0.8, and 0.6 nM, respectively. BI-882370 also inhibits SRC family kinases .
|
-

- HY-120181
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
AGI-14100 is a metabolically stable and orally available mIDH1 inhibitor (IC50=6 nM). The pharmacochemical optimization of AGI-14100 is aimed at eliminating hPXR activation, resulting in the final drug candidate AG-120. AG-120 can be used in the study of cancers carrying IDH1 mutations. The discovery and development of AGI-14100 can be used for further studies of mutant isocitrate dehydrogenase 1 (mIDH1) inhibitors .
|
-

- HY-134050
-
|
Apt-1
|
RIP kinase
Autophagy
Apoptosis
Beclin1
Necroptosis
|
Inflammation/Immunology
|
|
Apostatin-1 (Apt-1) is a potent TRADD inhibitor. Apostatin-1 can bind with TRADD-N (KD=2.17 μM), disrupting its binding to both TRADD-C and TRAF2. Apostatin-1 modulates the ubiquitination of RIPK1 and beclin 1. Apostatin-1 blocks apoptosis and restores cellular homeostasis by activating autophagy in cells with accumulated mutant tau, α-synuclein, or huntingtin .
|
-

- HY-120667
-
|
|
MDM-2/p53
Apoptosis
|
Cancer
|
|
DS-5272 is an orally acitve inhibitor for p53-MDM2 with an IC50 of 20 nM. DS-5272 inhibits the proliferation of SJSA-1 (wildtype p53, IC50=0.17 μM) and DLD-1 (mutant p53). DS-5272 arrest the cell cycle, and induces apoptosis in SJSA-1. DS-5272 exhibits antitumor efficacy in mice .
|
-

- HY-173277
-
|
|
Bacterial
|
Infection
|
|
FtsZ-IN-13 (Compound C11) is an inhibitor of temperature-sensitive mutant Z (FtsZ), with IC50 values of 47.97, 34 μM against FtsZSa and FtsZPa, respectively. FtsZ-IN-13 has a notable antimicrobial activity against S. aureus (minimum inhibitory concentration value of 2 μg/mL), cystic fibrosis S. aureus clinical isolates, and methicillin-resistant S. aureus (MRSA) strains. FtsZ-IN-13 can be used for antimicrobial resistance study .
|
-

- HY-101562
-
|
GDC-0077; RG6114
|
PI3K
Apoptosis
|
Cancer
|
|
Inavolisib (GDC-0077) is a potent, orally active, and selective PI3Kα inhibitor (IC50=0.038 nM). Inavolisib exerts its activity by binding to the ATP binding site of PI3K, thereby inhibiting the phosphorylation of PIP2 to PIP3. Inavolisib is more selective for mutant versus wild-type PI3Kα. Inavolisib can be used for the study of breast cancer .
|
-

- HY-17396R
-
|
KP363 Hydrochloride (Standard)
|
Reference Standards
Fungal
Antibiotic
|
Infection
|
|
Rilpivirine (hydrochloride) (Standard) is the analytical standard of Rilpivirine (hydrochloride). This product is intended for research and analytical applications. Rilpivirine (R278474) hydrochloride is a potent and specific diarylpyrimidine (DAPY) non-nucleoside reverse transcriptase inhibitor (NNRTI). Rilpivirine hydrochloride has high antiviral activity against wild-type HIV (EC50=0.4 nM) and mutant viruses (EC50=0.1-2.0 nM). Rilpivirine hydrochloride has a high genetic barrier to resistance development of HIV .
|
-

- HY-110088
-
|
|
MDM-2/p53
|
Cancer
|
|
SCH529074 is a potent and orally active p53 activator. SCH529074 binds specifically and conformation-dependently to p53 DBD ( DNA binding domain) with a Ki of 1-2 μM in a saturable manner. SCH529074 restores mutant p53 function and interrupts HDM2-mediated ubiquitination of wild Type p53. SCH529074 can be used for the study of non-small-cell lung carcinoma (NSCLC) .
|
-

- HY-E70719
-
|
|
FGFR
|
Cancer
|
|
FGFR3 activating mutations are drivers of malignancy in several human tissues, including bladder, lung, cervix, and blood. FGFR3 K650E is a mutant of FGFR3 that may be present in multiple myeloma cell lines. FGFR3 K650E Recombinant Human Active Protein Kinase is a recombinant FGFR3 K650E protein that can be used to study FGFR3 K650E-related functions .
|
-

- HY-E70759
-
|
|
p38 MAPK
|
Cancer
|
|
The mitogen-activated protein kinase kinase 6 (MKK6) is one of the upstream activators of p38 MAPK. MKK6 activates myocardial cell NF-κB and inhibits apoptosis in a p38 MAPK-dependent manner. MKK6 SDTD is a mutant of MKK6. MKK6 SDTD Recombinant Human Active Protein Kinase is a recombinant MKK6 SDTD protein that can be used to study MKK6 SDTD-related functions .
|
-

- HY-112301R
-
|
BLU-667 (Standard)
|
Reference Standards
RET
|
Cancer
|
|
Pralsetinib (Standard) is the analytical standard of Pralsetinib. This product is intended for research and analytical applications. Pralsetinib (BLU-667) is a highly potent, selective RET inhibitor. Pralsetinib (BLU-667) inhibits WT RET, RET mutants V804L, V804M, M918T and CCDC6-RET fusion with IC50s of 0.4, 0.3, 0.4, 0.4, and 0.4 nM, respectively .
|
-

- HY-112929B
-
|
|
Phosphatase
|
Cancer
|
|
(1S,2S,3R)-DT-061 is an enantiomer of DT-061 (HY-112929). DT-061 is an orally active activator of protein phosphatase 2A (PP2A). (1S,2S,3R)-DT-061 can be used as a negative control in the research of KRAS-mutant and MYC-driven lung cancer tumorigenesis .
|
-

- HY-144069
-
|
|
Trk Receptor
Apoptosis
|
Cancer
|
|
Pan-Trk-IN-3 (Compound 11g) is a potent inhibitor of pan-Trk and their drug-resistant mutants with IC50 values of 2, 3, 2, 21, 26, 5, 7 and 6 nM against TrkA, TrkB, TrkC, TrkA G595R, TrkA G667C, TrkA G667S, TrkA F589L and TrkC G623R, respectively. Pan-Trk-IN-3 displays excellent antitumor activity and induces apoptosis .
|
-

- HY-P10436
-
|
|
Raf
|
Cancer
|
|
Braftide is an allosteric inhibitor for BRAF kinase by targeting the dimer interface of BRAF kinase and inhibiting the formation of BRAF dimers. Braftide inhibits wild-type BRAF and oncogenic BRAF G469A with IC50 of 364 nM and 172 nM, respectively. Braftide inhibits MAPK signaling pathway, inhibits proliferation of KRAS mutant tumor cells (EC50 is 7.1 and 6.6 μM, for HCT116 and HCT-15), in combination of TAT sequence. Braftide can be used for cancer research .
|
-

- HY-173409
-
|
|
Androgen Receptor
Ser/Thr Protease
|
Inflammation/Immunology
|
|
AR antagonist 11 (Compound c2) is a selective AR antagonist with an IC50 of 0.019 μM. AR antagonist 11 is also effective against AR F877L/T878A mutant (IC50: 1.03 μM). AR antagonist 11 inhibits LNCaP cell proliferation and reduces PSA protein expression (IC50: 0.54 μM). AR antagonist 11 can be used for research of prostate cancer (PCa) .
|
-

- HY-18705
-
|
|
Apoptosis
Reactive Oxygen Species (ROS)
Caspase
Bcl-2 Family
Mitochondrial Metabolism
|
Endocrinology
|
|
Azoramide is a potent, orally active small-molecule modulator of the unfolded protein response (UPR). Azoramide improves ER protein folding and elevates ER chaperone capacity, which together protects cells against ER stress. Azoramide alleviates PLA2G6 mutant-induced ER stress through modulating unfolded protein response, and enhances the CERB signaling to rescue mitochondrial function, thereby preventing apoptosis of DA neurons. Azoramide has antidiabetic activity .
|
-

- HY-101561
-
|
BLU-285
|
c-Kit
PDGFR
|
Cancer
|
|
Avapritinib (BLU-285) is a highly potent, selective, and orally active KIT and PDGFRA activation loop mutant kinases inhibitor with IC50s of 0.27 and 0.24 nM for KIT D816V and PDGFRA D842V, respectively. Avapritinib (BLU-285) binds the active conformation of the kinase and shows antitumor activity. Avapritinib (BLU-285) attenuates the transport function of both ABCB1 and ABCG2 .
|
-

- HY-50703
-
MK-2461
1 Publications Verification
|
c-Met/HGFR
|
Cancer
|
|
MK-2461 is an ATP-competitive, selective and orally active wild-type and mutant c-Met inhibitor (IC50s: 0.4-2.5 nM). MK-2461 also inhibits Ron (IC50 of 7 nM) and Flt1 (IC50 of 10 nM), MK-2461 shows selective for c-MET over other kinases (lC50s = 22-7800 nM). MK-2461 can be used for the study of cancer, such as gastric cancer .
|
-

- HY-156103
-
|
|
Huntingtin
|
Neurological Disease
|
|
mHTT-IN-2 (compound 27) is a potent inhibitor (EC50=0.066 μM) of mutant huntingtin (mHTT). mHTT-IN-2 reduces canonical splicing of HTT RNA exons [49-50] and is a splicing regulator of the huntingtin (HTT) gene. mHTT-IN-2 exhibits inhibitory activity in vitro and in vivo in human HD stem cells and mouse BACHD models. mHTT-IN-2 may be used in the study of branaplam-related peripheral neuropathy .
|
-

- HY-131003
-
|
DS-6051b; AB-106; IBI-344
|
ROS Kinase
|
Cancer
|
|
Taletrectinib (DS-6051b) is a potent, orally active, and next-generation selective ROS1/NTRK inhibitor. Taletrectinib potently inhibits recombinant ROS1, NTRK1, NTRK2, and NTRK3 with IC50s of 0.207, 0.622, 2.28, and 0.98 nM, respectively. Taletrectinib also inhibits ROS1 G2032R and other Crizotinib-resistant ROS1 mutants .
|
-

- HY-E70582
-
|
|
Adenosine Deaminase
|
Others
|
|
E.coli tRNA adenosine deaminase is derived from E.coli and is an adenine deaminase that can deaminate adenine in single-stranded RNA (ssRNA, mainly the loop region within tRNA) or double-stranded RNA (dsRNA), but has no deamination activity on DNA. E.coli tRNA adenosine deaminase is a protein-modified mutant of adenine deaminase, which can efficiently deaminate adenine in ssDNA and can be applied to adenine base editors (ABE) and RNA m6A methylation sequencing .
|
-

- HY-114436
-
|
|
Ras
|
Cancer
|
|
MRTX-1257 is a selective, irreversible, covalent and orally active KRAS G12C inhibitor, with an IC50 of 900 pM for KRAS dependent ERK phosphorylation in H358 cells .
|
-

- HY-114436S
-
|
|
Isotope-Labeled Compounds
|
Cancer
|
|
MRTX-1257-d6 is the deuterium labeled MRTX-1257 (HY-114436). MRTX-1257 is a selective, irreversible, covalent and orally active KRAS G12C inhibitor, with an IC50 of 900 pM for KRAS dependent ERK phosphorylation in H358 cells .
|
-

- HY-153202
-
|
|
MDM-2/p53
Apoptosis
|
Cancer
|
|
SLMP53-2 is a mutant p53 reactivator. SLMP53-2 restores wild-type-like conformation and DNA-binding ability of mutp53-Y220C by enhancing its interaction with the Hsp70, leading to the reestablishment of p53 transcriptional activity. SLMP53-2 can induce cell cycle arrest, apoptosis and endoplasmic reticulum (ER) stress. SLMP53-2 exhibits antitumor activity .
|
-

- HY-E70777
-
|
|
RET
|
Cancer
|
|
Rearranged during transfection (RET) is a transmembrane receptor protein tyrosine kinase (PTK) activated normally by forming ternary complexes with its cognate ligands and co-receptors. RET alterations by point mutations and gene fusions were found in diverse cancers. RET V804E is a mutant of RET. RET V804E Recombinant Human Active Protein Kinase is a recombinant RET V804E protein that can be used to study RET V804E-related functions .
|
-

- HY-E70859
-
|
|
RET
|
Cancer
|
|
Rearranged during transfection (RET) is a transmembrane receptor protein tyrosine kinase (PTK) activated normally by forming ternary complexes with its cognate ligands and co-receptors. RET alterations by point mutations and gene fusions were found in diverse cancers. RET L790F is a mutant of RET. RET L790F Recombinant Human Active Protein Kinase is a recombinant RET L790F protein that can be used to study RET L790F-related functions .
|
-

- HY-162577
-
|
|
FGFR
|
Cancer
|
|
FGFR-IN-14 (compound 10h) is a pan-FGFR inhibitor. FGFR-IN-14 inhibits FGFR1, FGFR2, FGFR3 and FGFR2 V564F gatekeeper mutant with IC50s of 46, 41, 99, and 62 nM, respectively. FGFR-IN-14 strongly suppresses NCI-H520 lung cancer cells, SNU-16 and KATO III gastric cancer cells proliferation with IC50s of 19, 59, and 73 nM, respectively .
|
-

- HY-153341
-
|
CFT1946
|
PROTACs
Raf
|
Cancer
|
|
Tagarafdeg (CFT1946) is an orally active, CRBN-based and mutant-selective bifunctional degradation activating compound (BiDAC) degrader of BRAF V600E with a DC50 of 14 nM in A375 cells. Tagarafdeg is capable of degrading BRAF V600E (Class I), G469A (Class II), G466V (Class III) mutations, and the p61-BRAF V600E splice variant. Tagarafdeg can be used in tumor research .
|
-

- HY-E70771
-
|
|
RET
|
Cancer
|
|
Rearranged during transfection (RET) is a transmembrane receptor protein tyrosine kinase (PTK) activated normally by forming ternary complexes with its cognate ligands and co-receptors. RET alterations by point mutations and gene fusions were found in diverse cancers. RET L730I is a mutant of RET. RET L730I Recombinant Human Active Protein Kinase is a recombinant RET L730I protein that can be used to study RET L730I-related functions .
|
-

- HY-131033
-
|
|
Biochemical Assay Reagents
|
Others
|
|
L-Azidonorleucine hydrochloride, an unnatural amino acid, is A Methionine surrogate. L-Azidonorleucine hydrochloride can be used to label mammalian cell proteins and identify a diverse set of methionyl-tRNA synthetase (MetRS) mutants . L-Azidonorleucine (hydrochloride) is a click chemistry reagent, it contains an Azide group and can undergo copper-catalyzed azide-alkyne cycloaddition reaction (CuAAc) with molecules containing Alkyne groups. It can also undergo strain-promoted alkyne-azide cycloaddition (SPAAC) reactions with molecules containing DBCO or BCN groups.
|
-

- HY-170598
-
|
|
PROTACs
SARS-CoV
Virus Protease
|
Infection
|
|
HP211206 is a SARS-CoV-2 main protease (M pro) PROTAC degrader. HP211206 effectively degrades SARS-CoV-2 M pro and its drug-resistant mutants. HP211206 has an IC50 of 181.9 nM and a DC50 of 621 nM for M pro. HP211206 has antiviral activity. (Pink: H117 (HY-170599); Black: linker (HY-B0236); Blue: E3 ligase ligand (HY-41547)) .
|
-

- HY-E70779
-
|
|
RET
|
Cancer
|
|
Rearranged during transfection (RET) is a transmembrane receptor protein tyrosine kinase (PTK) activated normally by forming ternary complexes with its cognate ligands and co-receptors. RET alterations by point mutations and gene fusions were found in diverse cancers. RET V804M is a mutant of RET. RET V804M Recombinant Human Active Protein Kinase is a recombinant RET V804M protein that can be used to study RET V804M-related functions .
|
-

- HY-14588S1
-
|
|
Isotope-Labeled Compounds
HIV
HIV Protease
SARS-CoV
|
Infection
|
|
Lopinavir-d8 (ABT-378-d8) is the deuterium labeled Lopinavir. Lopinavir (ABT-378) is a highly potent, selective peptidomimetic inhibitor of the HIV-1 protease, with Kis of 1.3 to 3.6 pM for wild-type and mutant HIV protease. Lopinavir acts by arresting maturation of HIV-1 thereby blocking its infectivity . Lopinavir is also a SARS-CoV 3CLpro inhibitor with an IC50 of 14.2 μM .
|
-

- HY-E70767
-
|
|
RET
|
Cancer
|
|
Rearranged during transfection (RET) is a transmembrane receptor protein tyrosine kinase (PTK) activated normally by forming ternary complexes with its cognate ligands and co-receptors. RET alterations by point mutations and gene fusions were found in diverse cancers. RET G691S is a mutant of RET. RET G691S Recombinant Human Active Protein Kinase is a recombinant RET G691S protein that can be used to study RET G691S-related functions .
|
-

- HY-104066A
-
|
Xiliertinib tartrate; HMPL-309 tartrate
|
EGFR
|
Cancer
|
|
Theliatinib (Xiliertinib) tartrate is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib (tartrate) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-E70768
-
|
|
RET
|
Cancer
|
|
Rearranged during transfection (RET) is a transmembrane receptor protein tyrosine kinase (PTK) activated normally by forming ternary complexes with its cognate ligands and co-receptors. RET alterations by point mutations and gene fusions were found in diverse cancers. RET G810C is a mutant of RET. RET G810C Recombinant Human Active Protein Kinase is a recombinant RET G810C protein that can be used to study RET G810C-related functions .
|
-

- HY-E70773
-
|
|
RET
|
Cancer
|
|
Rearranged during transfection (RET) is a transmembrane receptor protein tyrosine kinase (PTK) activated normally by forming ternary complexes with its cognate ligands and co-receptors. RET alterations by point mutations and gene fusions were found in diverse cancers. RET M918T is a mutant of RET. RET M918T Recombinant Human Active Protein Kinase is a recombinant RET M918T protein that can be used to study RET M918T-related functions .
|
-

- HY-147405
-
|
PF-07284890; ARRY-461
|
Raf
ERK
|
Cancer
|
|
Tinlorafenib (PF-07284890) is the orally active inhibitor for BRAF and CRAF with IC50s of 5.8 nM and 4.1 nM. Tinlorafenib (Compound 10) inhibits V600E mutated BRAF and V600K mutanted BRAF with IC50s of 4.25 nM and 2.7 nM. Tinlorafenib can cross the blood brain barrier. Tinlorafenib demonstrates CNS penetration and can be used in the research of BRAF-associated malignant and benign tumors of the CNS as well as extracranial malignancies .
|
-

- HY-15164R
-
|
|
EGFR
|
Cancer
|
|
Icotinib (Hydrochloride) (Standard) is the analytical standard of Icotinib (Hydrochloride). This product is intended for research and analytical applications. Icotinib Hydrochloride (BPI-2009) is a potent and specific EGFR inhibitor with an IC50 of 5 nM; also inhibits mutant EGFRL858R, EGFRL858R/T790M, EGFRT790M and EGFRL861Q. Icotinib (Hydrochloride) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-E70775
-
|
|
RET
|
Cancer
|
|
Rearranged during transfection (RET) is a transmembrane receptor protein tyrosine kinase (PTK) activated normally by forming ternary complexes with its cognate ligands and co-receptors. RET alterations by point mutations and gene fusions were found in diverse cancers. RET R813Q is a mutant of RET. RET R813Q Recombinant Human Active Protein Kinase is a recombinant RET R813Q protein that can be used to study RET R813Q-related functions .
|
-

- HY-162253
-
|
|
HIV
|
Infection
|
|
HIV-1 inhibitor-64 (Compound 7c) is a wild-type HIV-1 inhibitor that effectively suppresses the activity of HIV-1 mutants E138K/Q148K and G140S/Q148R with EC50 values of 62.5 nM and 11.3 nM, respectively. HIV-1 inhibitor-64 exhibits antiviral activity and can be used in the research of AIDS .
|
-

- HY-E70722
-
|
|
FGFR
|
Cancer
|
|
FGFR4 is a transcriptional target of the PAX3-FOXO1 fusion gene. FGFR4 expression is significantly higher in rhabdomyosarcoma. FGFR4 V550E is a mutant of FGFR3 that may be present in rhabdomyosarcoma. FGFR4 V550E Recombinant Human Active Protein Kinase is a recombinant FGFR4 V550E protein that can be used to study FGFR4 V550EK-related functions .
|
-

- HY-13811
-
|
|
E1/E2/E3 Enzyme
Apoptosis
|
Cancer
|
|
NSC697923 is a potent UBE2N (ubiquitin-conjugating enzyme E2 N, Ubc13) inhibitor. NSC697923 induces neuroblastoma (NB) cell death via promoting nuclear importation of p53 in p53 wild-type NB cells. NSC697923 also induces cell death in p53 mutant NB cells by activation of JNK-mediated apoptotic pathway. NSC697923 inhibits DNA damage and NF-κB signaling. Antitumor activity .
|
-

- HY-DY1024
-
|
|
Huntingtin
Parasite
|
Infection
Neurological Disease
|
TPE-MI (Tetraphenylethene maleimide) (solution) is a thiol probe for measuring unfolded protein load and proteostasis in cells (the excitation wavelength is 350 nm and the emission wavelength is 470 nm). TPE-MI can report imbalances in proteostasis in induced pluripotent stem cell models of Huntington disease, as well as cells transfected with mutant Huntington exon 1 before the formation of visible aggregates. TPE-MI also detects protein damage following dihydroartemisinin research of the malaria parasitesPlasmodium falciparum .
Solution Concentration: 10 mM
|
-

- HY-14588R
-
|
ABT-378 (Standard)
|
Reference Standards
HIV
HIV Protease
SARS-CoV
|
Infection
Cancer
|
|
Lopinavir (Standard) is the analytical standard of Lopinavir. This product is intended for research and analytical applications. Lopinavir (ABT-378) is a highly potent, selective peptidomimetic inhibitor of the HIV-1 protease, with Kis of 1.3 to 3.6 pM for wild-type and mutant HIV protease. Lopinavir acts by arresting maturation of HIV-1 thereby blocking its infectivity . Lopinavir is also a SARS-CoV 3CL pro inhibitor with an IC50 of 14.2 μM .
|
-

- HY-19719A
-
|
ARQ-092 hydrochloride
|
Akt
Parasite
|
Infection
Cancer
|
|
Miransertib hydrochloride (ARQ-092 hydrochloride) is a potent, orally active, selective and allosteric Akt inhibitor with IC50s of 2.7 nM, 14 nM and 8.1 nM for Akt1, Akt2, Akt3, respectively. Miransertib hydrochloride is also a potent the AKT1-E17K mutant protein inhibitor and has the potential for PI3K/AKT-driven tumors and Proteus syndrome research . Miransertib hydrochloride is effective against Leishmania .
|
-

- HY-144269
-
|
|
Raf
|
Cancer
|
|
SHR902275 is a potent, selective, and orally active RAF inhibitor targeting RAS mutant cancers. SHR902275 has IC50s of 1.6 nM, 10 nM, and 5.7 nM for cRAF, bRAF wt, and bRAF V600E, respectively. SHR902275 shows cell growth inhibition with GI50s of 1.5 and 0.17 nM, 0.4 nM and 0.32 nM for H358, A375, Calu6, and SK-MEL2 cells respectively .
|
-

- HY-174427
-
|
|
LRRK2
|
Neurological Disease
|
|
RN341 is a specific type II kinase inhibitor of LRRK2 (IC50: 296 nM). RN341 inhibits LRRK2 phosphorylation and avoids S935 dephosphorylation by stabilizing the open conformation. RN341 rescues LRRK2-mediated kinesin motility block by preventing microtubule binding. RN341 effectively inhibits LRRK2 wild-type and G2019S mutant at the cellular level. RN341 provides a new direction for Parkinson's disease research.
|
-

- HY-169386
-
|
|
AUTOTACs
|
Cancer
|
|
YT 6-2 analog-1 (compound 2-3) is the p62/SQSTM1 targeting, autophagy-targeting ligand (ATL) of AUTOTAC ATC-324 (HY-169385), which is an bivalent AR (Androgen Receptor) degrader. ATC-324 can reduce nuclear AR levels and downregulate target gene expression of AR and AR-v7, and also has a degradation effect on common AR mutants in PCa .
|
-

- HY-19719
-
|
ARQ-092
|
Akt
Parasite
|
Infection
Cancer
|
|
Miransertib (ARQ-092) is a potent, orally active, selective and allosteric Akt inhibitor with IC50s of 2.7 nM, 14 nM and 8.1 nM for Akt1, Akt2, Akt3, respectively. Miransertib is also a potent the AKT1-E17K mutant protein inhibitor and has the potential for PI3K/AKT-driven tumors and Proteus syndrome research . Miransertib is effective against Leishmania .
|
-

- HY-172408
-
|
|
Reverse Transcriptase
HIV
|
Infection
|
|
NNRT-IN-6 (Compound 13a) is the non-nucleoside reverse transcriptase inhibitor (NNRT) for HIV-1 reverse transcriptase (HIV-1 RT) with IC50 of 0.41 μM. NNRT-IN-6 inhibits HIV-1 wildtype and mutant strains L100I, K103N, Y181C, Y188L, E138K, F227L/V106A and RES056 with EC50 of 6.2-250 nM .
|
-

- HY-14183
-
|
RSD1235 hydrochloride
|
Potassium Channel
|
Cardiovascular Disease
|
|
Vernakalant hydrochloride is a mixed voltage- and frequency-dependent Na + and atria-preferred K + channel blocker. IC50 for block by Vernakalant of wild-type and mutant Kv1.5 channels Fractional block is 13.35±0.93 μM, 0.61±0.03 μM, and 1.63±0.09 μM for Kv1.5 channel wt, Kv1.5 channel I508F, Kv1.5 channel T479A, respectively.
|
-

- HY-W015399
-
|
|
Fungal
|
Infection
|
|
4-Methylcinnamic acid is a cinnamic acid derivative with antifungal activity. 4-Methylcinnamic acid can overcome the tolerance of glutathione reductase mutants to 4-Methoxycinnamic acid (HY-N1387). When 4-Methylcinnamic acid is used in combination with traditional antifungal agents, it can reduce their minimum inhibitory concentrations. 4-Methylcinnamic acid can act as an intervention catalyst to enhance the efficacy of traditional antifungal agents and has application potential in targeting fungal cell wall disruption and controlling drug-resistant fungi .
|
-

- HY-170550
-
|
|
Ras
ERK
|
Cancer
|
|
KRAS G12C inhibitor 69 (Compound K09) is the inhibitor for mutant RAS protein KRASG12C with an IC50 of 4.36 nM. KRAS G12C inhibitor 69 inhibits the ERK phosphorylation in NCI-H358 and MIA-PACA-2 with an IC50 of 12 nM and 7 nM. KRAS G12C inhibitor 69 inhibits the proliferation of cancer cell NCI-H358 and MIA-PACA-2 with IC50 of 3.15 nM and 2.33 nM .
|
-

- HY-E70772
-
|
|
RET
|
Cancer
|
|
Rearranged during transfection (RET) is a transmembrane receptor protein tyrosine kinase (PTK) activated normally by forming ternary complexes with its cognate ligands and co-receptors. RET alterations by point mutations and gene fusions were found in diverse cancers. RET L730M is a mutant of RET. RET L730M Recombinant Human Active Protein Kinase is a recombinant RET L730M protein that can be used to study RET L730M-related functions .
|
-

- HY-157166
-
|
|
EGFR
|
Cancer
|
|
EGFR kinase inhibitor 2 (compound A-7) is a potent EGFR inhibitor targeting EGFR L858R/T790M/C797S and EGFR Del19/T790M/C797S mutants. EGFR kinase inhibitor 2 has the potential to address acquired resistance in the treatment of non-small cell lung cancer .
|
-

- HY-112611
-
|
|
Estrogen Receptor/ERR
|
Cancer
|
|
H3B-5942 is a selective, irreversible and orally active estrogen receptor covalent antagonist, inactivates both wild-type and mutant ERα by targeting Cys530, with Kis of 1 nM and 0.41 nM, respectively. H3B-5942 reduces ERα target gene GREB1, shows potent antitumor activity both in multiple cell lines or animals bearing ERα WT or ERα mutations .
|
-

- HY-E70774
-
|
|
RET
|
Cancer
|
|
Rearranged during transfection (RET) is a transmembrane receptor protein tyrosine kinase (PTK) activated normally by forming ternary complexes with its cognate ligands and co-receptors. RET alterations by point mutations and gene fusions were found in diverse cancers. RET R749T is a mutant of RET. RET R749T Recombinant Human Active Protein Kinase is a recombinant RET R749T protein that can be used to study RET R749T-related functions .
|
-

- HY-E70769
-
|
|
RET
|
Cancer
|
|
Rearranged during transfection (RET) is a transmembrane receptor protein tyrosine kinase (PTK) activated normally by forming ternary complexes with its cognate ligands and co-receptors. RET alterations by point mutations and gene fusions were found in diverse cancers. RET G810R is a mutant of RET. RET G810R Recombinant Human Active Protein Kinase is a recombinant RET G810R protein that can be used to study RET G810R-related functions .
|
-

- HY-149695
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-91 (compound 9) is an orally available EGFR inhibitor with blood-brain barrier penetrability. EGFR-IN-91 inhibits EGFR L858R/C797S and EGFR exon 19del/C797S, inducing tumor regression in xenograft (PDX) mouse models. EGFR-IN-91 has the potential to inhibit localized and metastatic non-small cell lung cancer (NSCLC) driven by EGFR mutants .
|
-

- HY-132842
-
|
DZD9008
|
EGFR
Btk
|
Cancer
|
|
Sunvozertinib (DZD9008) is a potent ErbBs (EGFR, Her2, especially mutant forms) and BTK inhibitor. Sunvozertinib shows IC50s of 20.4, 20.4, 1.1, 7.5, and 80.4 nM for EGFR exon 20 NPH insertion, EGFR exon 20 ASV insertion, EGFR L858R and T790M mutations, and Her2 Exon20 YVMA, and EGFR WT A431, respectively (patent WO2019149164A1, example 52) .
|
-

- HY-150602
-
|
|
Proteasome
|
Neurological Disease
|
|
20S Proteasome activator 1 is a potent 20S proteasome activator with EC200 values of 0.3 μM, 0.7 μM and 1.8 μM for trypsin-like site, chymotrypsin-like site and caspase-like site. 20S Proteasome activator 1 translates well in a cellular system, preventing the accumulation of the pathogenic A53T mutant of α-synuclein. 20S Proteasome activator 1 can be used for researching neurodegenerative diseases .
|
-

- HY-E70766
-
|
|
RET
|
Cancer
|
|
Rearranged during transfection (RET) is a transmembrane receptor protein tyrosine kinase (PTK) activated normally by forming ternary complexes with its cognate ligands and co-receptors. RET alterations by point mutations and gene fusions were found in diverse cancers. RET E762Q is a mutant of RET. RET E762Q Recombinant Human Active Protein Kinase is a recombinant RET E762Q protein that can be used to study RET E762Q-related functions .
|
-

- HY-172140
-
|
|
Potassium Channel
|
Neurological Disease
|
|
Ebio3 is a selective potassium channel (KCNQ2) inhibitor with an IC50 of 1.2 nM. Ebio3 binds to the KCNQ2 channel through its hydrophobic tail, causing the S6 helix to move inward, which leads to the closure of the inner gate. The inhibitory effect of Ebio3 is also effective in pathogenic mutants of KCNQ2 (such as R75C and I238L), where it can inhibit outward currents by more than 80%. Ebio3 is expected to be used in the research of neurological diseases such as epilepsy .
|
-

- HY-146349
-
|
|
PROTACs
EGFR
Autophagy
|
Cancer
|
|
PROTAC EGFR degrader 4 is a potent PROTAC targeting mutant EGFR.PROTAC EGFR degrader 4 induces EGFR del19 and EGFR L858R/T790M degradation with DC50s of 0.51 and 126 nM, respectively. PROTAC EGFR degrader 4 significantly inhibits growth of HCC827 and H1975 cell lines with IC50s of 0.83 and 203.1 nM, respectively. Induced EGFR degradation is related to autophagy .
|
-

- HY-104066
-
|
Xiliertinib; HMPL-309
|
EGFR
|
Cancer
|
|
Theliatinib (Xiliertinib) is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-E70780
-
|
|
RET
|
Cancer
|
|
Rearranged during transfection (RET) is a transmembrane receptor protein tyrosine kinase (PTK) activated normally by forming ternary complexes with its cognate ligands and co-receptors. RET alterations by point mutations and gene fusions were found in diverse cancers. RET Y791F is a mutant of RET. RET Y791F Recombinant Human Active Protein Kinase is a recombinant RET Y791F protein that can be used to study RET Y791F-related functions .
|
-

- HY-E70781
-
|
|
RET
|
Cancer
|
|
Rearranged during transfection (RET) is a transmembrane receptor protein tyrosine kinase (PTK) activated normally by forming ternary complexes with its cognate ligands and co-receptors. RET alterations by point mutations and gene fusions were found in diverse cancers. RET Y806H is a mutant of RET. RET Y806H Recombinant Human Active Protein Kinase is a recombinant RET Y806H protein that can be used to study RET Y806H-related functions .
|
-

- HY-123824
-
|
|
Sodium Channel
|
Neurological Disease
|
|
GNE-0439 is a novel Nav1.7-selective inhibitor with IC50 of 0.34 uM and inhibits Nav1.5 with an IC50 of 38.3 μM. GNE-0439 inhibits mutant N1742K channels (IC50=0.37 uM) in membrane potential assays. GNE-0439 possesses a carboxylic acid group, binds outside of the channel pore, and is unique compared with known selective VSD4 binders .
|
-

- HY-W102456
-
|
L-4-Acetylphenylalanine
|
Biochemical Assay Reagents
Amino Acid Derivatives
|
Others
|
|
H-Phe(4-Ac)-OH (L-4-Acetylphenylalanine) is a keto-amino acid that can be converted from α-keto acids containing an acetyl group. H-Phe(4-Ac)-OH can be added to the amber position to form mutant Z-domain proteins. H-Phe(4-Ac)-OH is used as a functional amino acid in peptide modification to achieve chemical bonding between peptides and solid surfaces .
|
-

- HY-164056
-
|
|
EGFR
|
Cancer
|
|
EGFR T790M/L858R-IN-8 (compound 9) is a potent inhibitor of EGFR targeting mutant EGFR T790M/L858R with an IC50 of 56.8 μM. The anti-proliferative effect of EGFR T790M/L858R-IN-8 on cancer cell lines A549, A431, and NHI-H1975 is not significant .
|
-

- HY-170497
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-140 (Compound 31) is the inhibitor for EGFR, that inhibits EGFR wildtype and EGFR L858R/T790M/C797S mutant with Ki of 0.95 nM and 2.1 nM, and inhibits EGFR del19/T790M/C797S in Ba/F3 with an IC50 of 56.9 nM. EGFR-IN-140 exhibits antitumor efficacy in mouse model .
|
-

- HY-E70770
-
|
|
RET
|
Cancer
|
|
Rearranged during transfection (RET) is a transmembrane receptor protein tyrosine kinase (PTK) activated normally by forming ternary complexes with its cognate ligands and co-receptors. RET alterations by point mutations and gene fusions were found in diverse cancers. RET G810S is a mutant of RET. RET G810S Recombinant Human Active Protein Kinase is a recombinant RET G810S protein that can be used to study RET G810S-related functions .
|
-

- HY-15666
-
|
GZD824; HQP1351
|
Bcr-Abl
|
Cancer
|
|
Olverembatinib (GZD824) is a potent and orally active pan-Bcr-Abl inhibitor. Olverembatinib potently inhibits a broad spectrum of Bcr-Abl mutants. Olverembatinib strongly inhibits native Bcr-Abl and Bcr-Abl T315I with IC50s of 0.34 nM and 0.68 nM, respectively. Olverembatinib has antitumor activity . Olverembatinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-155356A
-
|
|
PROTACs
|
Cancer
|
|
YN14 mixture of diastereomers is the diastereomers of YN14 (HY-155356).
YN14 is a KRASG12C proteolysis targeting chimera (PROTAC). YN14 is highly potent and selective KRASG12C degrader and induces a stable KRASG12C: YN14: VHL ternary complex with low binding free energy (ΔG). YN14 has antiproliferative effects and significantly inhibits KRASG12C-mutant cancer cell growth .
|
-

- HY-E70721
-
|
|
FGFR
|
Cancer
|
|
FGFR4 is a transcriptional target of the PAX3-FOXO1 fusion gene. FGFR4 expression is significantly higher in rhabdomyosarcoma. FGFR3 K650E is a mutant of FGFR3 that may be present in rhabdomyosarcoma. FGFR4 N535K Recombinant Human Active Protein Kinase is a recombinant FGFR4 N535K protein that can be used to study FGFR4 N535K-related functions .
|
-

- HY-109142
-
|
AK0529; RO-0529
|
RSV
|
Infection
|
|
Ziresovir (AK0529;RO-0529) is a potent, selective, and orally bioavailable respiratory syncytial virus (RSV) fusion (F) protein (RSV F) protein inhibitor. Ziresovir shows anti-RSV activity (EC50=3 nM) and highlights pharmacokinetics in animal species .
|
-

- HY-122936
-
|
|
Histone Methyltransferase
Keap1-Nrf2
Sirtuin
|
Cardiovascular Disease
Cancer
|
|
Tanshindiol C is a S-adenosylmethionine-competitive EZH2 (Histone Methyltransferase) inhibitor with an IC50 of 0.55 μM for inhibiting the methyltransferase activity. Tanshindiol C is also an activator of both Nrf2 and Sirtuin 1 (Sirt1) in macrophages. Tanshindiol C possesses anti-cancer activity, and can be used for atherosclerosis research .
|
-

- HY-137497
-
|
|
Ras
Apoptosis
|
Cancer
|
|
KRAS inhibitor-9, a potent KRAS inhibitor (Kd=92 μM), blocks the formation of GTP-KRAS and downstream activation of KRAS. KRAS inhibitor-9 binds to KRAS G12D, KRAS G12C and KRAS Q61H protein with a moderate binding affinity. KRAS inhibitor-9 causes G2/M cell cycle arrest and induces apoptosis. KRAS inhibitor-9 selectively inhibits the proliferation of NSCLC cells with KRAS mutation but not normal lung cells .
|
-

- HY-145723A
-
|
|
FLT3
FGFR
|
Inflammation/Immunology
Cancer
|
|
MAX-40279 hydrochloride is a dual and orally active inhibitor of FLT3 kinase and FGFR kinase. MAX-40279 hydrochloride is also effective against the FLT3 mutants such as FLT3 D835Y, suggesting that it can overcome resistance to Quizartinib (HY-13001) and Sorafenib (HY-10201). MAX-40279 hydrochloride inhibits NDRG1 phosphorylation at Ser330 and suppresses endothelial-to-mesenchymal transition (EndMT). MAX-40279 hydrochloride can be used for the research of acute myelogenous leukemia (AML) .
|
-

- HY-12755
-
|
CID-2950007
|
Ras
Apoptosis
|
Cancer
|
|
ML141 (CID-2950007) is a potent, allosteric, selective and reversible non-competitive inhibitor of Cdc42 GTPase. ML141 inhibits Cdc42 wild type and Cdc42 Q61L mutant with EC50s of 2.1 and 2.6 μM, respectively. ML141 shows low micromolar potency and selectivity against other members of the Rho family of GTPases (Rac1, Rab2, Rab7). ML141 do not show cytotoxicity in multiple cell lines .
|
-

- HY-164365
-
|
|
PROTACs
Ras
|
Cancer
|
|
PROTAC K-Ras degrader-2 (compound 48) is a pan-KRAS-mutant PROTAC degrader with an IC50 of ≤200 nM for KRAS G12V/RAF1. PROTAC K-Ras degrader-2 degrades SW620 KRAS G12D with a DC50 of ≤200 nM. PROTAC K-Ras degrader-2 inhibits cell growth of SW620 3D cell with an IC50 of ≤20 nM. (Pink: KRAS inhibitor-30 (HY-164366)) .
|
-

- HY-145723
-
|
|
FLT3
FGFR
|
Inflammation/Immunology
Cancer
|
|
MAX-40279 is a dual and orally active inhibitor of FLT3 kinase and FGFR kinase. MAX-40279 is also effective against the FLT3 mutants such as FLT3 D835Y, suggesting that it can overcome resistance to Quizartinib (HY-13001) and Sorafenib (HY-10201). MAX-40279 inhibits NDRG1 phosphorylation at Ser330 and suppresses endothelial-to-mesenchymal transition (EndMT). MAX-40279 can be used for the research of acute myelogenous leukemia (AML) .
|
-

- HY-17499
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
EGFR-IN-12 is a 4,6-disubstituted pyrimidine and is a potent, ATP-competitive, irreversible and highly selective EGFR inhibitor with an IC50of 21 nM. EGFR-IN-12 also inhibits mutant EGFR L858R and EGFR L861Q with IC50s of 63 nM and 4 nM, respectively. EGFR-IN-12 displays strong selectivity for EGFR over HER4 (IC50 = 7640 nM) and a panel of 55 other kinases. EGFR-IN-12 induces cells apoptosis and has antitumor activity .
|
-

- HY-174887
-
|
|
Thyroid Hormone Receptor
AMPK
Acetyl-CoA Carboxylase
Mitochondrial Metabolism
|
Metabolic Disease
|
|
THR-β agonist 9 is a potent, selective, and His435 mutation-sensitive THR-β (EC50: 3.2 nM) agonist. THR-β agonist 9 has moderate selectivity (approximately 10-fold) and good activation capacity (EC50: 134.2 nM to 515.5 nM) for multiple His435 mutants (H435A, H435Y, and H435R). THR-β agonist 9 has the potential to be used in the study of dyslipidemia, metabolic dysfunction-associated steatohepatitis (MASH), or resistance to thyroid hormone (RTH) .
|
-

- HY-130122
-
|
|
Molecular Glues
PROTACs
Apoptosis
|
Cancer
|
|
MG-277, a molecular glue degrader, effectively induces degradation of a translation termination factor based on Cereblon E3 ligand, GSPT1, with a DC50 of 1.3 nM. MG-277 potently inhibits tumor cell growth in a p53-independent manner, with IC50s of 3.5 nM for RS4;11 cells and 3.4 nM for p53 mutant RS4;11/IRMI-2 cells, respectively. Anticancer activity .
|
-

- HY-131003A
-
|
DS-6051b free base; AB-106 free base; IBI-344 free base
|
ROS Kinase
|
Cancer
|
|
Taletrectinib (DS-6051b) free base is a potent, orally active, and next-generation selective ROS1/NTRK inhibitor. Taletrectinib free base potently inhibits recombinant ROS1, NTRK1, NTRK2, and NTRK3 with IC50s of 0.207, 0.622, 2.28, and 0.98 nM, respectively. Taletrectinib free base also inhibits ROS1 G2032R and other Crizotinib-resistant ROS1 mutants .
|
-

- HY-17598R
-
|
|
Oxidative Phosphorylation
Reference Standards
Parasite
p38 MAPK
Raf
Apoptosis
|
Infection
Cancer
|
|
Rafoxanide (Standard) is the analytical standard of Rafoxanide. This product is intended for research and analytical applications. Rafoxanide is a poent, orally active halogenated salicylaniline agent with antiparasitic activity. Rafoxanide interferes with energy metabolism in trematodes by uncoupling oxidative phosphorylation. Rafoxanide is also found to be a potent inhibitor of the BRAF V600E mutant protein, which is important in colorectal cancer. Rafoxanide can be used for the control of infestation with Hemonchus species or Fasciola species in sheep and cattle as well as Oestrus ovis in sheep. Rafoxanide can also be used for cancer research .
|
-

- HY-E70704
-
|
|
EGFR
|
Cancer
|
|
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR G719C Recombinant Human Active Protein Kinase is a recombinant EGFR G719C protein that can be used to study EGFR G719C-related functions .
|
-

- HY-178341
-
|
|
Reverse Transcriptase
HIV
|
Infection
|
|
NNRT-IN-13 is a potent non-nucleoside reverse transcriptase (NNRT) inhibitor that directly inhibiting HIV-1 reverse transcriptase (IC₅₀ = 0.25 μM). NNRT-IN-13 exhibits excellent antiviral activity against wild-type HIV-1 (EC₅₀ = 0.0046 μM) and a broad spectrum of drug-resistant mutants. NNRT-IN-13 exhibits favorable in vivo metabolic and safety profiles. NNRT-IN-13 can be used for human immunodeficiency virus (HIV) research .
|
-

- HY-E70706
-
|
|
EGFR
|
Cancer
|
|
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR L718Q Recombinant Human Active Protein Kinase is a recombinant EGFR L718Q protein that can be used to study EGFR L718Q-related functions .
|
-

- HY-175271
-
|
|
PROTACs
Trk Receptor
ERK
|
Cancer
|
|
JWJ-01-378 is a selective TRK PROTAC degrader. JWJ-01-378 efficiently degrades WT TRK and TPM3-TRKA fusion proteins and inhibits downstream pERK signaling, while showing limited efficacy against TRK inhibitor resistant mutants and ALK fusions. JWJ-01-378 potently suppresses cancer cell proliferation .Pink: TRK ligand (HY-160520); Blue: CRBN ligase ligand (HY-103596); Black: linker
|
-

- HY-148416
-
|
|
MDM-2/p53
|
Cancer
|
|
p53 Activator 7 is a p53 mutation Y220C (MDM-2/p53) activator with an EC50 of 104 nM. p53 Activator 7 can bind to p53 mutant and restore its ability to bind DNA (WO2022213975A1; Example B-1) . p53 Activator 7 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-175667
-
|
|
HIV
Reverse Transcriptase
|
Infection
|
|
NNRT-IN-12 (compound 19) is a potent non-nucleoside reverse transcriptase (NNRT) inhibitor with an IC50 value of 0.107 μM for WT HIV-1 RT. NNRT-IN-12 shows anti-HIV-1 activity with EC50 value of 14.1, 5.03, 7.64, 27.1 nM for L100I, K103N, Y181C, Y188L mutant HIV-1 strains, respectively. NNRT-IN-12 shows cytotoxicity .
|
-

- HY-135805
-
|
|
EGFR
|
Cancer
|
|
JBJ-04-125-02 is a potent, mutant-selective, allosteric and orally active EGFR inhibitor with an IC50 of 0.26 nM for EGFR L858R/T790M. JBJ-04-125-02 can inhibit cancer cell proliferation and EGFR L858R/T790M/C797S signaling. JBJ-04-125-02 has anti-tumor activities .
|
-

- HY-137458A
-
|
ARQ 751 trihydrochloride
|
Akt
|
Cancer
|
|
Vevorisertib (ARQ 751) trihydrochloride is a selective, allosteric, pan-AKT and AKT1-E17K mutant inhibitors. Vevorisertib trihydrochloride potently inhibit phosphorylation of AKT. Vevorisertib trihydrochloride has Kd values of 1.2 nM and 8.6 nM for AKT1 and AKT1-E17K, respectively. Vevorisertib trihydrochloride has IC50 values of 0.55, 0.81, and 1.3 nM for AKT1, AKT2, and AKT3, respectively. Vevorisertib trihydrochloride can be used for the research of cancer .
|
-

- HY-151903S
-
|
|
FGFR
|
Cancer
|
|
FGFR2/3-IN-1 is a potent and selective FGFR2 and FGFR3 (FGFR) inhibitor with IC50s of 1 nM and 0.5 nM, respectively. FGFR2/3-IN-1 displays >40-fold selectivity over FGFR1/FGFR4 and other kinome. FGFR2/3-IN-1 also inhibits FGFR3 V555L and V555M mutants with IC50s of 2.7 nM and 6.1 nM, respectively .
|
-

- HY-16936
-
|
|
LRRK2
|
Neurological Disease
|
|
JH-II-127 is an orally active, highly potent, selective and brain-permeable LRRK2 inhibitor, with IC50s of 6, 2 and 48 nM for wild-type LRRK2 and LRRK2-G2019S and mutant LRRK2-A2016T. JH-II-127 inhibits Ser935 phosphorylation in all tissues of mice, including the brain. JH-II-127 can be used in the study of parkinson's syndrome .
|
-

- HY-170951
-
|
|
Androgen Receptor
|
Cancer
|
AR antagonist 10 (Compound Y5) is potent and orally active androgen receptor (AR) antagonist with an IC50 of 0.04 μM. AR antagonist 10 demonstrates dual mechanisms of action, antagonizes AR by disrupting AR dimerization, and induces AR degradation via the ubiquitin-proteasome pathway. AR antagonist 10 exhibits excellent activity against variant drug-resistant AR mutants. AR antagonist 10 effectively suppresses the tumor growth of the LNCaP xenograft. AR antagonist 10 is potential to be used for drug-resistant prostate cancer .
|
-

- HY-E70695
-
|
|
EGFR
|
Cancer
|
|
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR C797S Recombinant Human Active Protein Kinase is a recombinant EGFR C797S protein that can be used to study EGFR C797S-related functions .
|
-

- HY-E70707
-
|
|
EGFR
|
Cancer
|
|
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR L858R Recombinant Human Active Protein Kinase is a recombinant EGFR L858R protein that can be used to study EGFR L858R-related functions .
|
-

- HY-114365
-
|
UDP-N-acetyl-D-galactosamine disodium
|
Endogenous Metabolite
|
Metabolic Disease
Cancer
|
|
UDP-GalNAc (UDP-N-acetyl-D-galactosamine) disodium is a sugar nucleotide and a substrate for EpsC115. EpsC115 is a mutant with N-terminal residues 1-115 deleted from the exopolymeric substance (EPS). UDP-GalNAc disodium is a donor substrate for many N-acetylgalactosaminyltransferases, which transfer GalNAc from nucleotide sugars to sugar or peptide acceptors. UDP-GalNAc disodium provides a sugar group donor for glycosylation reactions. UDP-GalNAc disodium can be used in cancer research, such as colorectal and breast cancer .
|
-

- HY-128522
-
|
|
Ras
|
Cancer
|
|
ARS-1323-alkyne is a covalent inhibitor probe that covalently binds to the Switch-II pocket (S-IIP) of the KRAS G12C mutant protein. ARS-1323-alkyne visualizes the covalent modification of KRAS G12C and quantitatively measures the binding efficiency of the inhibitor to the target. ARS-1323-alkyne can be used to validate the target occupancy of KRAS G12C inhibitors and the synergistic mechanism of combination therapy, providing tool support for the development of combination treatment strategies .
|
-

- HY-126247
-
|
|
Ras
|
Cancer
|
|
BI-2852 is a KRAS inhibitor for the switch I/II pocket (SI/II-pocket) by structure-based agent design with nanomolar affinity. BI-2852 is mechanistically distinct from covalent KRASG12C inhibitor (binds to switch II pocket) and binds ten-fold more strongly to active KRASG12D versus KRASwt (740 nM vs 7.5 μM). BI-2852 blocks GEF, GAP, and effector interactions with KRAS, leading to inhibition of downstream signaling and an antiproliferative effect in KRAS mutant cells.
|
-

- HY-E70708
-
|
|
EGFR
|
Cancer
|
|
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR L861Q Recombinant Human Active Protein Kinase is a recombinant EGFR L861Q protein that can be used to study EGFR L861Q-related functions .
|
-

- HY-163393
-
|
|
Influenza Virus
|
Infection
|
|
Neuraminidase-IN-18 (compound N5) is a novel polyheterocyclic neuraminidase (NA) inhibitor. Neuraminidase-IN-18 shows potency in inhibition of H5N1 NA with an IC50 of 0.14 μM and 0.27 μM against the wild-type H5N1 NA and H5N1-H274Y mutant NA, respectively. Neuraminidase-IN-18 inhibits influenza virus replication by binding to NAs in cell level .
|
-

- HY-116191
-
|
|
PI3K
mTOR
|
Cancer
|
|
WJD008 is a potent dual phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) inhibitor with antiproliferative and anticlonogenic activity in tumor cells and transformed cells with PIK3CA mutant. WJD008 inhibits kinase activity of PI3K α and mTOR and abrogates insulin-like growth factor-I-activated PI3K-Akt-mTOR signaling cascade. WJD008 is promising for research of cancers .
|
-

- HY-130241
-
|
|
HIV
|
Infection
|
|
Reverse transcriptase-IN-1 (Compound 12z), a diarylbenzopyrimidine (DABP) analogue, is a potent, orally active HIV-1 nonnucleoside reverse transcriptase inhibitor. Reverse transcriptase-IN-1 has antiviral activity with EC50 values of 3.4 nM, 4.3 nM and 3.6 nM for HIV-1 IIIB, E138K and K103N mutants, respectively. Reverse transcriptase-IN-1 also has an IC50of 13.7 nM against HIV-1 reverse transcriptase enzyme .
|
-

- HY-176269
-
|
|
Bcr-Abl
ERK
STAT
PARP
Caspase
Apoptosis
|
Cancer
|
|
VS1150 (Compound 11) is a BCR-ABL-targeting phosphorylation-inducing chimeric small molecule (PHICS). VS1150 significantly inhibits oncogenic kinase BCR-ABL signaling by inducing inhibitory phosphorylation at its Y253 (EC50: 69 nM), subsequently triggering cell apoptosis. VS1150 also inhibits other oncogenic ABL fusions and drug-resistant mutants like T315I. VS1150 can be used for chronic myeloid leukemia (CML) and other ABL fusion-driven cancers research .
|
-

- HY-15164AR
-
|
BPI-2009 (Standard)
|
Reference Standards
EGFR
|
Cancer
|
|
Icotinib (Standard) is the analytical standard of Icotinib. This product is intended for research and analytical applications. Icotinib (BPI-2009) is a potent and specific EGFR inhibitor with an IC50 of 5 nM; also inhibits mutant EGFR L858R, EGFR L858R/T790M, EGFR T790M and EGFR L861Q. Icotinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-P99934
-
|
ABBV-621
|
Apoptosis
Caspase
TNF Receptor
|
Cancer
|
|
Eftozanermin alfa (ABBV-621) is a tumor necrosis factor-related apoptosis-inducing ligand receptor (TRAIL-R) agonist. Eftozanermin alfa is a fusion protein consisting of a mutant immunoglobulin G1-Fc linked to 2 single-chain trimers of TRAIL. Eftozanermin alfa induces apoptosis in tumor cells by activation of death receptors (DR4 receptor and DR5 receptor) with Kds of 780 nM and 635 nM. Eftozanermin alfa can be used for the research of multiple solid and heme malignancies .
|
-

- HY-169387
-
|
|
AUTOTACs
|
Cancer
|
|
YT 6-2-PEG3-C2-NH2 is the p62/SQSTM1 targeting, autophagy-targeting ligand-linker conjugate of AUTOTAC ATC-324 (HY-169385), which is an bivalent AR (Androgen Receptor) degrader. ATC-324 can reduce nuclear AR levels and downregulate target gene expression of AR and AR-v7, and also has a degradation effect on common AR mutants in PCa .
|
-

- HY-P5395
-
|
|
HIV
|
Others
|
|
TAT-GluR23A Fusion Peptide is a biological active peptide. (This is the GluR23A sequence, a control inactive peptide used as a mutant counterpart to glutamate receptor endocytosis inhibitor (GluR23Y), connected to an 11 amino acid cell permeable HIV Trans-Activator of Transcription (TAT) protein transduction domain (PTD). GluR23A is derived from GluR23Y amino acids 869 to 877, with Ala substituted for Tyr, and thus lacking essential phosphorylation sites.Control peptide of HY-P2259)
|
-

- HY-15666A
-
|
GZD824 dimesylate; HQP1351 dimesylate
|
Bcr-Abl
|
Cancer
|
|
Olverembatinib (GZD824) dimesylate is a potent and orally active pan-Bcr-Abl inhibitor. Olverembatinib dimesylate potently inhibits a broad spectrum of Bcr-Abl mutants. Olverembatinib dimesylate strongly inhibits native Bcr-Abl and Bcr-Abl T315I with IC50s of 0.34 nM and 0.68 nM, respectively. Olverembatinib dimesylate has antitumor activity . Olverembatinib (dimesylate) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-164474
-
|
|
MEK
|
Cancer
|
|
DS03090629 is an orally active MEK inhibitor that inhibits MEK activity in an ATP-competitive manner. DS03090629 exhibits high affinity for both MEK and phosphorylated MEK, with Kd values of 0.11 and 0.15 nM, respectively. It effectively inhibits the proliferation of BRAF-mutant overexpressing melanoma cell lines, with IC50 values of 74.3 and 97.8 nM for BRAF V600E and MEK1 F53L transfected A375 cells, respectively. DS03090629 holds potential value in the field of anti-melanoma therapy .
|
-

- HY-111545
-
|
|
FLT3
|
Cancer
|
|
BSc5371 is a potent and irreversible FLT3 inhibitor, with Kds of 1.3, 0.83, 1.5, 5.8 and 2.3 nM for mutant FLT3(D835H), FLT3(ITD, D835V), FLT3(ITD, F691L), FLT3-ITD and wild type FLT3wt, respectively. BSc5371 is cytotoxic to FLT3-dependent cell lines .
|
-

- HY-101561R
-
|
BLU-285 (Standard)
|
Reference Standards
c-Kit
PDGFR
|
Cancer
|
|
Avapritinib (Standard) is the analytical standard of Avapritinib. This product is intended for research and analytical applications. Avapritinib (BLU-285) is a highly potent, selective, and orally active KIT and PDGFRA activation loop mutant kinases inhibitor with IC50s of 0.27 and 0.24 nM for KIT D816V and PDGFRA D842V, respectively. Avapritinib (BLU-285) binds the active conformation of the kinase and shows antitumor activity. Avapritinib (BLU-285) attenuates the transport function of both ABCB1 and ABCG2 .
|
-

- HY-E70742
-
|
|
c-Kit
|
Cancer
|
|
KIT (CD117) is an important cell surface marker used to identify certain types of hematopoietic(blood) progenitors in the bone marrow. KIT is a cytokine receptor expressed on the surface of hematopoietic stem cells as well as other cell types. Altered forms of this receptor may be associated with some types of cancer. KIT V654A is a mutant of KIT. KIT V654A Recombinant Human Active Protein Kinase is a recombinant KIT V654A protein that can be used to study KIT V654A-related functions .
|
-

- HY-E70705
-
|
|
EGFR
|
Cancer
|
|
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR G719S Recombinant Human Active Protein Kinase is a recombinant EGFR G719S protein that can be used to study EGFR G719S-related functions .
|
-

- HY-155059
-
|
|
Bacterial
|
Infection
|
|
Antimycobacterial agent-6 (compound 25) is a potent inhibitor of Mycobacterium tuberculosis (Mtb),targeting to both wild-type and fluoroquinolone-resistant Mtb strains. Antimycobacterial agent-6 inhibits Mtb DprE1-C387S mutant with MIC90s of 0.9 μM (H37Rv),0.9 μM (MoxR),0.5 μM (DprE1-P116S),respectively .
|
-

- HY-174442
-
|
|
PARP
|
Cancer
|
|
PARP1-IN-40 is a highly selectively and orally active PARP1 inhibitor (IC50: 0.19 nM for PARP1, 26 nM for PARP2). PARP1-IN-40 kills tumor cells by inhibiting PARP1, leading to accumulation of DNA damage. PARP1-IN-40 has high antitumor activity against BRCA mutant MDA-MB-436 cells. PARP1-IN-40 can be used in combination with chemotherapy for cancer-related research .
|
-

- HY-E70709
-
|
|
EGFR
|
Cancer
|
|
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR T790M Recombinant Human Active Protein Kinase is a recombinant EEGFR T790M protein that can be used to study EGFR T790M-related functions .
|
-

- HY-174261
-
|
|
Ras
|
Cancer
|
|
KRAS-IN-5 (Compound Ex 6) is an orally active and selective inhibitor targeting KRAS mutants (including KRAS G12D, KRAS G12V, KRAS WT) with a GNE IC50 value of 1.3 nM against KRAS G12D. KRAS-IN-5 blocks tumor cell proliferation by inhibiting KRAS-mediated signaling pathways (e.g., reducing ERK phosphorylation). KRAS-IN-5 is promising for research of KRAS mutation-related cancers, such as pancreatic cancer, colorectal cancer, lung cancer .
|
-

- HY-E70738
-
|
|
c-Kit
|
Cancer
|
|
KIT (CD117) is an important cell surface marker used to identify certain types of hematopoietic(blood) progenitors in the bone marrow. KIT is a cytokine receptor expressed on the surface of hematopoietic stem cells as well as other cell types. Altered forms of this receptor may be associated with some types of cancer. KIT V559D is a mutant of KIT. KIT V559D Recombinant Human Active Protein Kinase is a recombinant KIT V559D protein that can be used to study KIT V559D-related functions .
|
-

- HY-171835
-
|
|
HIV
HIV Protease
|
Infection
|
|
DPC 684 is a potent and selective HIV-1 protease inhibitor (IC90 = 5.7-40 nM, Ki = 0.021 nM). DPC 684 competitively inhibits HIV-1 protease and blocks viral polyprotein cleavage. DPC 684 has low protein binding and broad-spectrum inhibition against a variety of wild-type and mutant HIV-1 proteases. DPC 684 has low protein binding and broad-spectrum inhibition (IC90 = 1.9-6.3 nM). DPC 684 has research significance for HIV .
|
-

- HY-E70737
-
|
|
c-Kit
|
Cancer
|
|
KIT (CD117) is an important cell surface marker used to identify certain types of hematopoietic(blood) progenitors in the bone marrow. KIT is a cytokine receptor expressed on the surface of hematopoietic stem cells as well as other cell types. Altered forms of this receptor may be associated with some types of cancer. KIT T670I is a mutant of KIT. KIT T670I Recombinant Human Active Protein Kinase is a recombinant KIT T670I protein that can be used to study KIT T670I-related functions .
|
-

- HY-145723B
-
|
|
FLT3
FGFR
|
Inflammation/Immunology
Cancer
|
|
MAX-40279 hemifumarate is a dual and orally active inhibitor of FLT3 kinase and FGFR kinase. MAX-40279 hemifumarate is also effective against the FLT3 mutants such as FLT3 D835Y, suggesting that it can overcome resistance to Quizartinib (HY-13001) and Sorafenib (HY-10201). MAX-40279 hemifumarate inhibits NDRG1 phosphorylation at Ser330 and suppresses endothelial-to-mesenchymal transition (EndMT). MAX-40279 hemifumarate can be used for the research of acute myelogenous leukemia (AML) .
|
-

- HY-X0009
-
|
GFH009; JSH-009; SLS009
|
CDK
DYRK
Apoptosis
Bcl-2 Family
c-Myc
Caspase
PARP
DNA/RNA Synthesis
|
Metabolic Disease
Inflammation/Immunology
Cancer
|
|
Tambiciclib (GFH009, JSH-009) is an orally active, highly potent and selective CDK9 inhibitor (IC50 = 1 nM), demonstrating >200-fold selectivity over other CDKs, >100-fold selectivity over DYRK1A/B, and excellent selectivity over 468 kinases/mutants. Tambiciclib demonstrates potent in vitro and in vivo antileukemic efficacy in acute myeloid leukemia (AML) mouse models by inhibiting RNA Pol II phosphorylation, downregulating MCL1 and MYC, and inducing apoptosis. Tambiciclib can be used for AML research .
|
-

- HY-114323
-
|
|
PROTACs
FLT3
Apoptosis
STAT
MEK
ERK
|
Cancer
|
|
PROTAC FLT-3 degrader 1 is an effective and selective FLT-3 PROTAC degrader with an IC50 of 0.6 nM. PROTAC FLT-3 degrader 1 inhibits both FLT-3 and FLT-3 ITD mutants. PROTAC FLT-3 degrader 1 has the activity of anti-proliferation and induction of apoptosis, which can be used in the study of tumor. (Pink: FLT3 ligand (HY-168702); Black: Linker (HY-124380); Blue: VHL ligand (HY-125845)) .
|
-

- HY-E70703
-
|
|
EGFR
|
Cancer
|
|
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR d752-759 Recombinant Human Active Protein Kinase is a recombinant EGFR d752-759 protein that can be used to study EGFR d752-759-related functions .
|
-

- HY-15164AS
-
|
BPI-2009-d4
|
Isotope-Labeled Compounds
|
Others
|
|
Icotinib-d4 is the deuterium-labeled Icotinib (HY-15164A). Icotinib-d4 (BPI-2009) is a potent and specific EGFR inhibitor with an IC50 of 5 nM; also inhibits mutant EGFR L858R, EGFR L858R/T790M, EGFR T790M and EGFR L861Q. Icotinib-d4 is a click chemistry reagent, itcontains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-15666R
-
|
GZD824 (Standard); HQP1351 (Standard)
|
Reference Standards
Bcr-Abl
|
Cancer
|
|
Olverembatinib (Standard) is the analytical standard of Olverembatinib. This product is intended for research and analytical applications. Olverembatinib (GZD824) is a potent and orally active pan-Bcr-Abl inhibitor. Olverembatinib potently inhibits a broad spectrum of Bcr-Abl mutants. Olverembatinib strongly inhibits native Bcr-Abl and Bcr-Abl T315I with IC50s of 0.34 nM and 0.68 nM, respectively. Olverembatinib has antitumor activity . Olverembatinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-148597
-
|
|
FGFR
Cytochrome P450
|
Cancer
|
|
FGFR-IN-10 is an orally active inhibitor of FGFR and Cytochrome P450 (CYPs). FGFR-IN-10 inhibits wide type and V564F mutant FGFR2 with IC50s of 104.1 nM and 43.6 nM, respectively. FGFR-IN-10 also inhibits CYPs with IC50s of 3.33 μM (CYP2C9), 18.75 μM (CYP2C19), 4.34 μM (CYP2CD6), and 0.69 μM (CYP3A4), respectively .
|
-

- HY-W754911
-
|
BLU-285-d3
|
Isotope-Labeled Compounds
PDGFR
c-Kit
|
Cancer
|
|
Avapritinib-d3 (BLU-285-d3) is deuterium labeled Avapritinib. Avapritinib (BLU-285) is a highly potent, selective, and orally active KIT and PDGFRA activation loop mutant kinases inhibitor with IC50s of 0.27 and 0.24 nM for KIT D816V and PDGFRA D842V, respectively. Avapritinib (BLU-285) binds the active conformation of the kinase and shows antitumor activity. Avapritinib (BLU-285) attenuates the transport function of both ABCB1 and ABCG2 .
|
-

- HY-162720
-
|
|
HIV
Potassium Channel
Reverse Transcriptase
|
Infection
|
|
NNRT-IN-4 (Compound 10p) is an inhibitor for non-nucleoside reverse transcriptase (NNRT) with an IC50 of 0.713 µM for HIV-1 RT. NNRT-IN-4 exhibits antiviral efficacy, inhibits HIV-1 wildtype and mutant strains with EC50 of 6-63 nM. NNRT-IN-4 exhibits a slight inhibitory activities against hERG (IC50=25.9 µM) and CYP enzymes (IC50>50 µM). NNRT-IN-4 exhibits good tolerability and safety in mice (2 g/kg) .
|
-

- HY-112289
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
IDH889 is an orally available, brain penetrant, allosteric and mutant specific inhibitor of isocitrate dehydrogenase 1 (IDH1). IDH889 has potent selectivity for IDH1 R132* mutations, with IC50s of 0.02 μM, 0.072 μM and 1.38 μM for IDH1 R132H, IDH1 R132C and IDH1 wt, respectively. IDH889 shows potent cellular inhibition of R-2-hydroxyglutarate (2-HG) production with an IC50 of 0.014 μM .
|
-

- HY-P11357A
-
|
|
IFNAR
|
Cancer
|
|
KRAS G12C Peptide TFA is the trifluoroacetate salt of KRAS G12C Peptide (HY-P11357). KRAS G12C Peptide is a specific peptide derived from the Kirsten rat sarcoma virus (KRAS) gene carrying the G12C oncogenic mutation. KRAS G12C Peptide induces responses like IFN-γ secretion and cytotoxicity. KRAS G12C Peptide can be used for the study of immune responses against KRAS G12C-mutant tumors .
|
-

- HY-134850
-
|
|
PROTACs
Tau Protein
|
Neurological Disease
|
|
QC-01-175 is a heterobifunctional molecule, which degrades aberrant tau. QC-01-175 reduces the levels of A152T and P301L mutant tau protein and protects neurons from tau-mediated toxicity and improve cell survival (Pink: ligand for target protein Aberrant tau ligand 1 (HY-W453397); Black: linker NH2-PEG3 (HY-W007545); Blue: ligand for E3 ligase Pomalidomide (HY-10984)) .
|
-

- HY-155356
-
|
|
PROTACs
Ras
|
Cancer
|
|
YN14 is a KRASG12C proteolysis targeting chimera (PROTAC). YN14 is highly potent and selective KRASG12C degrader and induces a stable KRASG12C: YN14: VHL ternary complex with low binding free energy (ΔG). YN14 has antiproliferative effects and significantly inhibits KRASG12C-mutant cancer cell growth. YN14 leads to tumor regression with tumor growth inhibition (TGI%) rates more than 100 % in the MIA PaCa-2 xenograft model.
|
-

- HY-157403
-
|
|
Virus Protease
SARS-CoV
|
Infection
|
|
Jun12682 is an orally active SARS-CoV-2 papain-like protease (PL pro) inhibitor, with a Ki value of 37.7 nM and an EC50 value of 1.1 μM in the FlipGFP PL pro assay. Jun12682 has efficacy in hindering PL pro both deubiquitination and deISGylation, with Ki values of 63.5 and 38.5 nM, respectively. Jun12682 exhibits resistance in multiple PL pro mutant strains, and its enzymatic activity is comparable to that of the wild-type. Jun12682 can be used for the study of the SARS-CoV-2 .
|
-

- HY-170799
-
|
|
DNA/RNA Synthesis
SARS-CoV
Arenavirus
|
Infection
|
|
HNC-1664 is the orally active inhibitor for RNA-dependent RNA polymerase (RdRP). HNC-1664 exhibits broad-spectrum antiviral activity against coronavirus (SARS-CoV-2 wildtype and its mutants XBB.1.18, HK.3.1, BF.7.14, BA.1HCoV-229E, HCoV-OC43) and arenavirus. HNC-1664 exhibits anti-infectious activity in SARS-CoV-2 Delta infected mouse models .
|
-

- HY-164373
-
|
|
Androgen Receptor
Apoptosis
|
Cancer
|
|
SC428 is an androgen receptor (AR) inhibitor that targets the N-terminal domain. SC428 potently decrease the transactivation of (AR)-V7, (AR)v567es, as well as full-length ( AR ) (AR-FL) and its LBD mutants, substantially. SC428 inhibits androgen-stimulated (AR)-FL nuclear translocation, chromatin binding, and (AR) -regulated gene transcription. SC428 inhibits the proliferation of tumor cells in vitro. SC428 inhibits tumor cell growth by inducing apoptosis in mice transplanted with 22RV1 .
|
-

- HY-E70697
-
|
|
EGFR
|
Cancer
|
|
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR d746-750 Recombinant Human Active Protein Kinase is a recombinant EGFR d746-750 protein that can be used to study EGFR d746-750-related functions .
|
-

- HY-169349
-
|
|
Androgen Receptor
|
Cancer
|
|
Androgen receptor antagonist 12 (Compound EF2) is an orally active Androgen receptor (AR) antagonist (IC50: 0.30 μM). Androgen receptor antagonist 12 inhibits transcriptional activity of variant AR mutants and and the proliferation of AR-positive PCa cell lines. Androgen receptor antagonist 12 blocks AR nuclear translocation. Androgen receptor antagonist 12 inhibits tumor growth in a C4-2B xenograft mouse model. Androgen receptor antagonist 12 can be used for prostate cancer (PCa) research .
|
-

- HY-50707
-
|
(Rac)-T-Type calcium channel inhibitor
|
Calcium Channel
Drug Isomer
|
Neurological Disease
|
|
(Rac)-TTA-P2 is the isomer of TTA-P2 (HY-10035), and can be used as an experimental control. TTA-P2 (T-Type calcium channel inhibitor) is a potent inhibitor of T-Type calcium channel. TTA-P2 penetrates well the CNS and blocks the native T-type currents in deep cerebellar nuclear neurons, the window current is completely abolished both for wild-type and mutant Cav3.1 channels. TTA-P2 has the potential for the research of neurology disease .
|
-

- HY-E70741
-
|
|
c-Kit
|
Cancer
|
|
KIT (CD117) is an important cell surface marker used to identify certain types of hematopoietic(blood) progenitors in the bone marrow. KIT is a cytokine receptor expressed on the surface of hematopoietic stem cells as well as other cell types. Altered forms of this receptor may be associated with some types of cancer. KIT V560G is a mutant of KIT. KIT V560G Recombinant Human Active Protein Kinase is a recombinant KIT V560G protein that can be used to study KIT V560G-related functions .
|
-

- HY-14588S
-
|
(rel)-ABT-378-d8
|
Isotope-Labeled Compounds
HIV Protease
SARS-CoV
HIV
|
Others
|
|
(rel)-Lopinavir-d8 ((rel)-ABT-378-d8) is the deuterium labeled Lopinavir (HY-14588) . Lopinavir (ABT-378) is a highly potent, selective peptidomimetic inhibitor of the HIV-1 protease, with Kis of 1.3 to 3.6 pM for wild-type and mutant HIV protease. Lopinavir acts by arresting maturation of HIV-1 thereby blocking its infectivity . Lopinavir is also a SARS-CoV 3CL pro inhibitor with an IC50 of 14.2 μM .
|
-

- HY-E70736
-
|
|
c-Kit
|
Cancer
|
|
KIT (CD117) is an important cell surface marker used to identify certain types of hematopoietic(blood) progenitors in the bone marrow. KIT is a cytokine receptor expressed on the surface of hematopoietic stem cells as well as other cell types. Altered forms of this receptor may be associated with some types of cancer. KIT D816V is a mutant of KIT. KIT D816V Recombinant Human Active Protein Kinase is a recombinant KIT D816V protein that can be used to study KIT D816V-related functions .
|
-

- HY-137460
-
|
K0706
|
Bcr-Abl
|
Cancer
|
|
Vodobatinib (K0706) is a potent, third generation and orally active Bcr-Abl1 tyrosine kinase inhibitor with an IC50 of 7 nM. Vodobatinib exhibits activity against most BCR-ABL1 point mutants, and has no activity against BCR-ABL1T315I. Vodobatinib can be used for chronic myeloid leukemia (CML) research . Vodobatinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-146223
-
|
|
Ras
p38 MAPK
PI3K
Apoptosis
|
Cancer
|
|
AZD4625 is an orally active, selective irreversible, covalent allosteric GTPase KRASG12C inhibitor with an IC50 of 3 nM. AZD4625 can inhibit the MAPK pathway (with decreased pCRAF, pMEK, and pERK) and the PI3K pathway (with decreased pAKT and pS6), and induce cell apoptosis. AZD4625 has no binding and inhibition of wild-type RAS or isoforms carrying non-KRASG12C mutations. AZD4625 can be used for the study of KRASG12C mutant non-small cell lung cancer .
|
-

- HY-E70735
-
|
|
c-Kit
|
Cancer
|
|
KIT (CD117) is an important cell surface marker used to identify certain types of hematopoietic(blood) progenitors in the bone marrow. KIT is a cytokine receptor expressed on the surface of hematopoietic stem cells as well as other cell types. Altered forms of this receptor may be associated with some types of cancer. KIT D816H is a mutant of KIT. KIT D816H Recombinant Human Active Protein Kinase is a recombinant KIT D816H protein that can be used to study KIT D816H-related functions .
|
-

- HY-E70714
-
|
|
Anaplastic lymphoma kinase (ALK)
|
Cancer
|
|
EML4-ALK is a fusion-type protein tyrosine kinase produced through gene rearrangement. EML4-ALK can be used for the study of non-small cell lung carcinoma (NSCLC) and neuroblastoma (NB). EML4-ALK has multiple mutants. EML4 ALK F1174L Recombinant Human Active Protein Kinase is a recombinant EML4 ALK F1174L protein that can be used to study EML4 ALK F1174L-related functions .
|
-

- HY-145723C
-
|
|
FLT3
FGFR
|
Inflammation/Immunology
Cancer
|
|
MAX-40279 hemiadipate is a dual and orally active inhibitor of FLT3 kinase and FGFR kinase. MAX-40279 hemiadipate is also effective against the FLT3 mutants such as FLT3 D835Y, suggesting that it can overcome resistance to Quizartinib (HY-13001) and Sorafenib (HY-10201). MAX-40279 hemiadipate inhibits NDRG1 phosphorylation at Ser330 and suppresses endothelial-to-mesenchymal transition (EndMT). MAX-40279 hemiadipate can be used for the research of acute myelogenous leukemia (AML) .
|
-

- HY-178148
-
|
|
Androgen Receptor
|
Endocrinology
Cancer
|
|
AR antagonist 17 is a selective, orally active, low brain-penetrant Androgen Receptor (AR) antagonist (IC50 = 0.010 μM), effectively blocking AR dimerization and nuclear translocation, and demonstrating potent efficacy in several castration-resistant prostate cancer (CRPC) cells. AR antagonist 17 showed superior efficacy against variant drug-resistant AR mutants. AR antagonist 17 can inhibit tumor growth in an LNCaP xenograft model without apparent toxicity. AR antagonist 17 can be used for the study of castration-resistant prostate cancer (CRPC) .
|
-

- HY-15814
-
|
|
Bcr-Abl
PDGFR
c-Kit
Src
JAK
Apoptosis
|
Cancer
|
|
HG-7-85-01 is a type II ATP competitive inhibitor of wild-type and gatekeeper mutations forms of Bcr-Abl, PDGFRα, Kit, and Src kinases. HG-7-85-01 inhibits T315I mutant Bcr-Abl kinase, KDR and RET with IC50s of 3 nM, 20 nM and 30 nM, and is only weak or no inhibition of other kinases (IC50>2 μM). HG-7-85-01 inhibits the cell proliferation, which is mediated by the induction of apoptosis, and inhibition of cell-cycle progression .
|
-

- HY-10035A
-
|
|
Calcium Channel
|
Neurological Disease
|
|
(R)-TTA-P2 is the isomer of TTA-P2 (HY-10035), and can be used as an experimental control. TTA-P2 (T-Type calcium channel inhibitor) is a potent inhibitor of T-Type calcium channel. TTA-P2 penetrates well the CNS and blocks the native T-type currents in deep cerebellar nuclear neurons, the window current is completely abolished both for wild-type and mutant Cav3.1 channels. TTA-P2 has the potential for the research of neurology disease .
|
-

- HY-E70718
-
|
|
FGFR
|
Cancer
|
|
FGFR3 activating mutations are drivers of malignancy in several human tissues, including bladder, lung, cervix, and blood. FGFR3 G697C is a mutant of FGFR3 that may be present in oral squamous cell carcinoma. FGFR3 G697C increases FGFR3 auto-phosphorylation. FGFR3 G697C Recombinant Human Active Protein Kinase is a recombinant FGFR3 G697C protein that can be used to study FGFR3 G697C-related functions .
|
-

- HY-136910
-
|
USP7-IN-7
|
Deubiquitinase
MDM-2/p53
|
Cancer
|
|
USP7-797 (USP7-IN-7) is an orally available, selective USP7 inhibitor (IC50=0.5 nmol/L) with antitumor activity. USP7-797 reduces the level of MDM2, thereby increasing the stability and activity of p53, leading to cell cycle arrest and apoptosis. USP7-797 has low nanomolar cytotoxicity against p53 mutant cancer cell lines, p53 wild-type hematological tumors, and neuroblastoma cell lines .
|
-

- HY-100818
-
|
TAS-120
|
FGFR
|
Cancer
|
|
Futibatinib (TAS-120) is an orally bioavailable, highly selective, and irreversible FGFR inhibitor, with IC50s of 3.9, 1.3, 1.6, and 8.3 nM for FGFR 1-4, respectively. Futibatinib inhibits mutant and wild-type FGFR2 with similar IC50s (wild-type FGFR2=0.9 nM; V5651=1-3 nM; N550H=3.6 nM; E566G=2.4 nM) .
|
-

- HY-P11356A
-
|
|
IFNAR
|
Cancer
|
|
KRAS G12V Peptide TFA is the trifluoroacetate salt of KRAS G12V Peptide (HY-P11355). KRAS G12V Peptide is a specific peptide derived from the Kirsten rat sarcoma virus (KRAS) gene carrying the G12V oncogenic mutation. KRAS G12V Peptide induces responses like IFN-γ secretion and cytotoxicity. KRAS G12V Peptide can be used for the study of immune responses against KRAS G12V-mutant tumors .
|
-

- HY-E70734
-
|
|
c-Kit
|
Cancer
|
|
KIT (CD117) is an important cell surface marker used to identify certain types of hematopoietic(blood) progenitors in the bone marrow. KIT is a cytokine receptor expressed on the surface of hematopoietic stem cells as well as other cell types. Altered forms of this receptor may be associated with some types of cancer. KIT A829P is a mutant of KIT. KIT A829P Recombinant Human Active Protein Kinase is a recombinant KIT A829P protein that can be used to study KIT A829P-related functions .
|
-

- HY-178501
-
|
|
Ras
|
Cancer
|
|
KRAS-IN-45 (Compound 1.019) is a KRAS inhibitor. KRAS-IN-45 can be used for the study of cancers .
|
-

- HY-130149
-
|
MRTX849
|
Ras
|
Cancer
|
|
Adagrasib (MRTX849) is a potent, orally-available, and mutation-selective covalent inhibitor of KRAS G12C with potential antineoplastic activity. Adagrasib covalently binds to KRAS G12C at the cysteine at residue 12, locks the protein in its inactive GDP-bound conformation, and inhibits KRAS-dependent signal transduction .
|
-
- HY-178502
-
|
|
Ras
|
Cancer
|
|
KRAS-IN-46 (Compound 1.013) is a KRAS inhibitor. KRAS-IN-46 can be used for the study of cancers .
|
-

- HY-E70754
-
|
|
c-Met/HGFR
|
Cancer
|
|
Mesenchymal-to-epithelial transition (MET) is a receptor tyrosine kinase for hepatocyte growth factor (HGF). MET overactivation is strongly associated with angiogenesis, cellular motility, growth, and invasion. Aberrant MET signaling can drive tumorigenesis in several cancer types through various molecular mechanisms, including MET amplification, MET exon 14 skipping mutation, MET overexpression, and MET fusions. MET Y1230A is a mutant of MET. MET Y1230A Recombinant Human Active Protein Kinase is a recombinant MET Y1230A protein that can be used to study MET Y1230A-related functions .
|
-

- HY-169076
-
|
|
FLT3
HDAC
Apoptosis
|
Cancer
|
FLT3/HDAC-IN-1 is a dual inhibitor of FLT3/HDAC, with IC50 values of 30.4, 52.4, and 14.7 nM for FLT3, HDAC1, and HDAC3, respectively. FLT3/HDAC-IN-1 can induce apoptosis in MV-4-11 cells and has anti-proliferative effects on FLT3 mutant-transformed BaF3 cells. FLT3/HDAC-IN-1 is being researched for its potential in treating hard-to-treat solid tumors and hematological malignancies .
|
-

- HY-163799
-
|
|
Histone Methyltransferase
|
Cancer
|
|
PRMT5-MTA-IN-1 (Compound A9a) is an inhibitor for protein arginine methyltransferase PRMT5-MTA. PRMT5-MTA-IN-1 inhibits the proliferation of colorectal cancer cell HCT116 wildtype and MTAP del mutant, with an IC50 of 16 nM and 2.47 μM. PRMT5-MTA-IN-1 exhibits good liver microsomal stability and film permeability. PRMT5-MTA-IN-1 exhibits good pharmacokinetic characteristics in CD-1 mice .
|
-

- HY-155302
-
|
|
FLT3
|
Cancer
|
|
FLT3-IN-22 (compound 22f) is a potent FLT3 inhibitor with IC50 values of 0.941 nM and 0.199 nM for FLT3 and FLT3/D835Y, respectively. FLT3-IN-22 exhibits strong antiproliferative activity against MV4-11 cells and the mutant FLT kinase expressed Ba/F3 cell lines, including FLT-D835Y and FLT3-F691L .
|
-

- HY-P10976
-
|
|
SARS-CoV
|
Infection
|
|
Spike Glycoprotein (1147-1162) is a linear and broadly neutralizing peptide in the S2 protein of SARS-CoV-2. Spike Glycoprotein (1147-1162) is conserved across SARS-CoV, BatCoV RaTG13, SARS-CoV-2, and SARS-CoV-2 variants. Spike Glycoprotein (1147-1162)-targeting mAbs can neutralize both SARS-CoV-2 and SARS-CoV by preventing fusion between the virus and cell membrane. Spike Glycoprotein (1147-1162) can be used for universal vaccines against SARS-CoV-2 mutants research .
|
-

- HY-164477
-
|
|
Androgen Receptor
|
Cancer
|
|
FL442 is an Androgen Receptor (AR) modulator. FL442 exhibits strong inhibitory effects in AR-dependent prostate cancer cells, showing similar inhibitory efficiency to traditional antiandrogen drugs Bicalutamide (HY-14249) and Enzalutamide (HY-70002), while maintaining antiandrogenic activity against the AR mutant F876L, which is highly resistant to Enzalutamide. Pharmacokinetic studies of FL442 in mice reveal a long half-life (8 hours), good targeting (prostate tissue), and metabolic stability, and it effectively inhibits LNCaP tumor growth at low plasma concentrations (30 ng/mL) .
|
-

- HY-X0009A
-
|
GFH009 dimaleate; JSH-009 dimaleate; SLS009 dimaleate
|
CDK
DYRK
Apoptosis
Bcl-2 Family
c-Myc
Caspase
PARP
DNA/RNA Synthesis
|
Cancer
|
|
Tambiciclib (GFH009, JSH-009) dimaleate is an orally active, highly potent and selective CDK9 inhibitor (IC50 = 1 nM), demonstrating >200-fold selectivity over other CDKs, >100-fold selectivity over DYRK1A/B, and excellent selectivity over 468 kinases/mutants. Tambiciclib dimaleate demonstrates potent in vitro and in vivo antileukemic efficacy in acute myeloid leukemia (AML) mouse models by inhibiting RNA Pol II phosphorylation, downregulating MCL1 and MYC, and inducing apoptosis. Tambiciclib dimaleate can be used for AML research .
|
-

- HY-178031
-
|
|
HIV
|
Infection
|
|
HIV-1-IN-87 (Compound 11x) is a dual-site HIV-1 inhibitor targeting the non-nucleoside reverse transcriptase inhibitor binding pocket (NNIBP) and its adjacent site (NNIAS). HIV-1-IN-87 has potent antiviral activities against wild-type and mutant strains (such as L100I, K103N and Y181C) (EC50s: 4.1-150 nM). HIV-1-IN-87 can be used for HIV-1 infections like acquired immune deficiency syndrome (AIDS) research .
|
-

- HY-156498
-
|
|
Ras
ERK
Raf
Ribosomal S6 Kinase (RSK)
AMPK
Apoptosis
PARP
|
Cancer
|
|
RMC-7977 is an orally active triple-complex RAS inhibitor that can simultaneously bind to cyclophilin A (CYPA) (Kd = 195 nM) and KRAS (G12V) (Kd = 292 μM). It exhibits broad-spectrum inhibitory activity against KRAS, NRAS, and HRAS proteins and their various wild-type and mutant variants. RMC-7977 induces apoptosis by inhibiting the phosphorylation of ERK, CRAF, and RSK, as well as increasing PARP cleavage. This leads to tumor regression, reduces resistance in KRAS G12C cancer models, and demonstrates good tolerability across various RAS cancer models .
|
-

- HY-N2099
-
|
Senegin III
|
Autophagy
Amyloid-β
Caspase
NF-κB
Apoptosis
|
Neurological Disease
Metabolic Disease
Inflammation/Immunology
|
|
Onjisaponin B is an orally active natural product derived from Polygala tenuifolia. Onjisaponin B inhibits NF-κB p65. Onjisaponin B enhances autophagy and accelerates the degradation of mutant α-synuclein and huntingtin. Onjisaponin B reduces β-amyloid (Aβ) production. Onjisaponin B reduces radiation-induced cell apoptosis. Onjisaponin B has anti-oxidant and anti-inflammatory activities. Onjisaponin B can be used for neurological disease and radiation injury study, and its metabolite tenuifolin (TF) can enter the brain through the BBB .
|
-

- HY-153857
-
|
PHI-101
|
FLT3
Checkpoint Kinase (Chk)
|
Cancer
|
|
Lasmotinib (PHI-101) is a FLT3 and CHK2 inhibitor. Lasmotinib potently inhibits FLT3 single activating mutations (ITD or TKD mutants) and has inhibitory activity against FLT3 double (ITD/D835Y or ITD/F691L) and triple (ITD/D835Y/F691L) resistance mutations. Lasmotinib synergizes with Venetoclax (HY-15531) or Azacytidine to inhibit leukemia. Lasmotinib exhibits anticancer activity against ovarian and breast cancer .
|
-

- HY-145269
-
|
|
Cadherin
|
Cancer
|
|
AL-GDa62 is a derivative of the CDH1/E-cadherin modulator SLEC-11 (HY-145268) and induces apoptosis in CDH1 -/- cells. AL-GDa62 has an EC50 of 3.2 μM and 2 μM for isogenic mammary epithelial cells MCF10A-WT (wild type) and mutant MCF10A-CDH1 -/-, respectively. AL-GDa62 specifically inhibits TCOF1, ARPC5, and UBC9, and suppresses SUMOylation at low micromolar concentrations .
|
-

- HY-158101
-
|
CC-94676
|
PROTACs
Androgen Receptor
|
Cancer
|
|
BMS-986365 (CC-94676) is an orally active and selective targeted androgen receptor (AR) PROTAC degrader, capable of inducing cereblon (CRBN) E3 ligase-dependent ubiquitination and degradation of the androgen receptor (AR), as well as various AR mutants. BMS-986365 shows significant in vivo potency, degrading AR, inhibiting AR signaling, and restricting tumor growth in animal models of advanced prostate cancer. (Blue: HY-W247437; Black: linker (HY-W126831); Pink: HY-168697) .
|
-

- HY-175144
-
|
|
Ligands for Target Protein for PROTAC
Ras
|
Cancer
|
|
KRASG12D-IN-6 is a PROTAC target protein ligand that can be used to synthesize CH091138 (HY-175025). CH091138 is a potent and selective KRASG12D PROTAC degrader with anti-tumor activity .
|
-

- HY-E70752
-
|
|
c-Met/HGFR
|
Cancer
|
|
Mesenchymal-to-epithelial transition (MET) is a receptor tyrosine kinase for hepatocyte growth factor (HGF). MET overactivation is strongly associated with angiogenesis, cellular motility, growth, and invasion. Aberrant MET signaling can drive tumorigenesis in several cancer types through various molecular mechanisms, including MET amplification, MET exon 14 skipping mutation, MET overexpression, and MET fusions. MET L1195V is a mutant of MET. MET L1195V Recombinant Human Active Protein Kinase is a recombinant MET L1195V protein that can be used to study MET L1195V-related functions .
|
-

- HY-168858
-
|
|
Trk Receptor
|
Cancer
|
|
TRK-IN-30 (Compound C11) is the inhibitor for tropomyosin receptor kinase (TRK) that inhibits TRKA, TRKB and TRKC and drug resistant mutant TRKA G595R with an IC50 of 1.8, 0.98, 3.8, and 54 nM, respectively. TRK-IN-30 inhibits the activation of the downstream PI3K/AKT and MEK/ERK signaling pathways. TRK-IN-30 inhibits the colony formation and cell migration of Km-12, arrests the cell cycle at G0/G1 phase, and induces apoptosis in Km-12 .
|
-

- HY-163299
-
|
|
Ras
Apoptosis
PI3K
Akt
p38 MAPK
|
Cancer
|
|
pan-KRAS-IN-5 is a pan-KRAS translation inhibitor by targeting 5′-UTR RNA G-quadruplexes (rG4s). pan-KRAS-IN-5 strongly binds to and stabilizes KRAS rG4s, inhibits KRAS translation, and blocks the MAPK and PI3K-AKT pathways. pan-KRAS-IN-5 induces cell cycle arrest, prompts apoptosis in KRAS-driven cancer cells. pan-KRAS-IN-5 inhibits tumor growth and KRAS expression in KRAS-mutant xenograft. KRAS-IN-5 can be used for KRAS-driven cancer research .
|
-

- HY-161536
-
|
|
PROTACs
EGFR
Apoptosis
|
Cancer
|
|
PROTAC EGFR degrader 9 (Compound C6) is an orally active CRBN-based PROTAC EGFR degrader. PROTAC EGFR degrader 9 exhibits a DC50 of 10.2 nM and a Kd of 240.2 nM against EGFR L858R/T790M/C797S. PROTAC EGFR degrader 9 exhibits potent degradation activity against various EGFR mutants, while sparing the EGFRWT. (Blue: CRBN ligand (HY-A0003), Black: linker (HY-161613); Pink: EGFR inhibitor (HY-161537)) .
|
-

- HY-176556
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-1671 is a selective EGFR inhibitor with an IC50 of 0.19 nM. EGFR-IN-167 exhibits good potency against various EGFR mutants (IC50 = 0.109 nM, 0.75 nM and <0.05 nM against EGFR (L858R), EGFR (C797S) and EGFR (del19), respectively). EGFR-IN-1671 covalently engages the catalytically conserved lysine of EGFR in live mammalian cells. EGFR-IN-1671 demonstrates excellent anti-proliferative activity by inhibiting EGFR autophosphorylation. EGFR-IN-1671 can be used for the study of non-small-cell lung carcinomas (NSCLC), glioblastoma and many solid tumors .
|
-

- HY-E70756
-
|
|
c-Met/HGFR
|
Cancer
|
|
Mesenchymal-to-epithelial transition (MET) is a receptor tyrosine kinase for hepatocyte growth factor (HGF). MET overactivation is strongly associated with angiogenesis, cellular motility, growth, and invasion. Aberrant MET signaling can drive tumorigenesis in several cancer types through various molecular mechanisms, including MET amplification, MET exon 14 skipping mutation, MET overexpression, and MET fusions. MET Y1230D is a mutant of MET. MET Y1230D Recombinant Human Active Protein Kinase is a recombinant MET Y1230D protein that can be used to study MET Y1230D-related functions .
|
-

- HY-150652
-
|
|
FGFR
Apoptosis
|
Cancer
|
|
FGFR-IN-8 (Compound 17a) is a highly potent and orally active panFGFR inhibitor against wild-type and mutant FGFRs. FGFR-IN-8 shows inhibition with IC50 values of <0.5, 189.1, <0.5, 22.6, <0.5 and 7.30 nM against FGFR1, V564F-FGFR2, N549H-FGFR2, V555M-FGFR3, FGFR3 and FGFR4, respectively. GFR-IN-8 induces cancer cell apoptosis and shows anticancer activities .
|
-

- HY-E70758
-
|
|
c-Met/HGFR
|
Cancer
|
|
Mesenchymal-to-epithelial transition (MET) is a receptor tyrosine kinase for hepatocyte growth factor (HGF). MET overactivation is strongly associated with angiogenesis, cellular motility, growth, and invasion. Aberrant MET signaling can drive tumorigenesis in several cancer types through various molecular mechanisms, including MET amplification, MET exon 14 skipping mutation, MET overexpression, and MET fusions. MET Y1235D is a mutant of MET. MET Y1235D Recombinant Human Active Protein Kinase is a recombinant MET Y1235D protein that can be used to study MET Y1235D-related functions .
|
-

- HY-150926
-
|
|
Ras
|
Cancer
|
|
G12Si-1 is a selective K-Ras(G12S) covalent inhibitor, which can inhibit oncogenic signaling of K-Ras(G12S). G12Si-1 shows good ability to covalently engage recombinant K-Ras(G12S) at the mutant serine residue. G12Si-1 can also affect nucleotide cycling of K-Ras by blocking Sos-catalyzed exchange and decreasing the rate of EDTA promoted exchange .
|
-

- HY-177554
-
|
|
Biochemical Assay Reagents
|
Cancer
|
|
KB05yne is a site-selective, alkyne-functionalized electrophilic "scout" fragment probe designed for covalent ligand discovery, with preferential reactivity toward specific cysteine residues in proteins. KB05yne strongly labels wild-type forms of target proteins (e.g., CDKN2AIP, HPCAL1) but not their cysteine-to-alanine mutants, confirming site-specific cysteine reactivity. KB05yne can be used for site-specific covalent ligand discovery and optimization for structurally and functionally diverse proteins, particularly in studies targeting cysteine residues in cancer-related or difficult-to-purify proteins .
|
-

- HY-164473
-
|
|
ERK
Akt
STAT
Apoptosis
|
Cancer
|
|
DETD-35 is an inhibitor of the MEK-ERK, Akt, and STAT3 signaling pathways, which promotes cancer cell apoptosis (Apoptosis) and reduces cancer cell resistance to Vemurafenib (HY-12057). The IC50 values of DETD-35 against wild-type and mutant melanoma cell lines B16-F10, MeWo, SK-MEL-2, A2058c, and A375c are 2.7, 6.0, 3.9, 3.1, and 2.5 μM, respectively. DETD-35 holds promise for research in the field of anti-melanoma therapy .
|
-

- HY-159190
-
|
|
MAPKAPK2 (MK2)
|
Cancer
|
|
HRX-0233 is a small-molecule MAP2K4 inhibitor. HRX-0233 results in strong tumor shrinkage without any apparent toxicity in H358 KRASG12C-mutant non-small cell lung cancers (NSCLC) in vivo. HRX-0233 efficiently prevents feedback activation of receptor tyrosine kinases (RTKs) upon monotherapy KRAS inhibitor Sotorasib (HY-114277) and causes a more sustained and complete inhibition of MAPK signaling. HRX-0233 is promising for research of AR-negative prostate cancer, lung and colon cancers .
|
-

- HY-174458
-
|
|
PROTACs
MDM-2/p53
IKZF Family
Casein Kinase
|
Cancer
|
|
MD-4251 is an orally active MDM2 PROTAC degrader. MD-4251 potently degrades MDM2 in RS4;11 cells (DC50: 0.2 nM) and actives p53. MD-4251 shows strong antiproliferative activity against acute leukemia cells (wild-type p53) with minimal efficacy in mutant type. MD-4251 induces complete tumor regression in RS4;11 xenograft mice model . Pink: MDM2 ligand (HY-130684); Blue: CRBN ligase ligand (HY-W883326); Black: linker
|
-

- HY-170640
-
|
|
FLT3
Reactive Oxygen Species (ROS)
Apoptosis
|
Cancer
|
|
FLT3-IN-29 (Compound MY-10) is a FLT3 inhibitor (IC50s: 6.5 and 10.3 nM for FLT3-ITD and FLT3-D835Y mutants). FLT3-IN-29 arrests cell cycle at the G0/G1 phase and efficiently induces Apoptosis. FLT3-IN-29 also reduces reactive oxygen species (ROS) production and mitochondrial membrane potential (MMP). FLT3-IN-29 displays antileukemic activity .
|
-

- HY-E70749
-
|
|
c-Met/HGFR
|
Cancer
|
|
Mesenchymal-to-epithelial transition (MET) is a receptor tyrosine kinase for hepatocyte growth factor (HGF). MET overactivation is strongly associated with angiogenesis, cellular motility, growth, and invasion. Aberrant MET signaling can drive tumorigenesis in several cancer types through various molecular mechanisms, including MET amplification, MET exon 14 skipping mutation, MET overexpression, and MET fusions. MET D1228N is a mutant of MET. MET D1228N Recombinant Human Active Protein Kinase is a recombinant MET D1228N protein that can be used to study MET D1228N-related functions .
|
-

- HY-E70748
-
|
|
c-Met/HGFR
|
Cancer
|
|
Mesenchymal-to-epithelial transition (MET) is a receptor tyrosine kinase for hepatocyte growth factor (HGF). MET overactivation is strongly associated with angiogenesis, cellular motility, growth, and invasion. Aberrant MET signaling can drive tumorigenesis in several cancer types through various molecular mechanisms, including MET amplification, MET exon 14 skipping mutation, MET overexpression, and MET fusions. MET D1228H is a mutant of MET. MET D1228H Recombinant Human Active Protein Kinase is a recombinant MET D1228H protein that can be used to study MET D1228H-related functions .
|
-

- HY-B0984A
-
|
|
Calcium Channel
Ras
STING
Autophagy
|
Infection
Cardiovascular Disease
Cancer
|
|
Fendiline, a diphenylalkylamine type of antianginal agent, is an L-type calcium channel blocker (IC50 of 17 µM). Fendiline is also a selective K-Ras inhibitor, and has no effect on H-Ras and N-Ras. Fendiline inhibits K-Ras plasma membrane localization (IC50 of 9.64 μM), inhibits K-Ras signal output and blocks the proliferation of pancreatic, colon, lung, and endometrial cancer cell lines expressing oncogenic mutant K-Ras. Fendiline is a STING agonist and is able to inhibit the growth of multiple refractory cold tumors (MC38, CT26 and B16F10) .
|
-

- HY-149127
-
|
ASC-JM17; ALZ-003
|
Keap1-Nrf2
Androgen Receptor
HSP
Mitophagy
|
Metabolic Disease
|
|
Rosolutamide (ASC-JM17), a curcumin analog, is an orally active, potent Nrf1 and Nrf2 activator. Rosolutamide activates Nrf1, Nrf2 and heat shock factor 1 (Hsf1), thereby activating the expression of proteasome subunits, antioxidant enzymes and molecular chaperones. Rosolutamide degrades the polyglutamine (polyQ) androgen receptor (AR) via the ubiquitin-proteasome pathway and improves motor function in mouse models of spinal and bulbar muscular atrophy (SBMA). Rosolutamide improves mitochondrial function and promotes autophagy, decreases mutant protein aggregates, and attenuates intracellular/mitochondrial reactive oxygen species (ROS) levels .
|
-

- HY-E70755
-
|
|
c-Met/HGFR
|
Cancer
|
|
Mesenchymal-to-epithelial transition (MET) is a receptor tyrosine kinase for hepatocyte growth factor (HGF). MET overactivation is strongly associated with angiogenesis, cellular motility, growth, and invasion. Aberrant MET signaling can drive tumorigenesis in several cancer types through various molecular mechanisms, including MET amplification, MET exon 14 skipping mutation, MET overexpression, and MET fusions. MET Y1230C is a mutant of MET. MET Y1230C Recombinant Human Active Protein Kinase is a recombinant MET Y1230C protein that can be used to study MET Y1230C-related functions .
|
-

- HY-13238
-
|
S/GSK1349572
|
HIV Integrase
HIV
|
Infection
|
|
Dolutegravir (S/GSK1349572) is a highly potent and orally bioavailable HIV integrase strand transfer inhibitor with an IC50 of 2.7 nM for HIV-1 integrase-catalyzed strand transfer. Dolutegravir (S/GSK1349572) inhibits HIV-1 viral replication with an IC50 of 0.51 nM in peripheral blood mononuclear cells. Dolutegravir retains a high potency against the HIV-1 Y143R, N155H, and G140S/Q148H mutants (EC50=3.6-5.8 nM) .
|
-

- HY-162702
-
|
|
PROTACs
Androgen Receptor
|
Cancer
|
|
AZ‘3137 is an orally active PROTAC-type androgen receptor (AR) degrader with a DC50 value of 22 nM. AZ‘3137 can degrade L702H mutant AR (DC50 of 92 nM). AZ'3137 can inhibit cell proliferation of LNCaP, with a GI50 value of 74 nM. AZ'3137 can inhibit AR signaling and tumor growth in prostate cancer mice (Pink: AR Ligand (HY-172954); Blue: CRBN ligand (HY-172955); Black: linker (HY-W262798); E3 Ligand+Linker: HY-172956) .
|
-

- HY-B0984
-
|
|
Calcium Channel
Ras
STING
Autophagy
|
Infection
Cardiovascular Disease
Cancer
|
|
Fendiline hydrochloride, a diphenylalkylamine type of antianginal agent, is an L-type calcium channel blocker (IC50 of 17 µM). Fendiline hydrochloride is also a selective K-Ras inhibitor, and has no effect on H-Ras and N-Ras. Fendiline hydrochloride inhibits K-Ras plasma membrane localization (IC50 of 9.64 μM), inhibits K-Ras signal output and blocks the proliferation of pancreatic, colon, lung, and endometrial cancer cell lines expressing oncogenic mutant K-Ras. Fendiline hydrochloride is a STING agonist and is able to inhibit the growth of multiple refractory cold tumors (MC38, CT26 and B16F10) .
|
-

- HY-100818R
-
|
TAS-120 (Standard)
|
Reference Standards
FGFR
|
Cancer
|
|
Futibatinib (Standard) is the analytical standard of Futibatinib. This product is intended for research and analytical applications. Futibatinib (TAS-120) is an orally bioavailable, highly selective, and irreversible FGFR inhibitor, with IC50s of 3.9, 1.3, 1.6, and 8.3 nM for FGFR 1-4, respectively. Futibatinib inhibits mutant and wild-type FGFR2 with similar IC50s (wild-type FGFR2=0.9 nM; V5651=1-3 nM; N550H=3.6 nM; E566G=2.4 nM) .
|
-

- HY-128131
-
|
|
Endogenous Metabolite
|
Others
|
|
UCCF-029 Free base is a potent activator of the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel. UCCF-029 Free base exhibits enhanced activity through benzannulation of the flavone A-ring at the 7,8-position. UCCF-029 Free base serves as a structural guide for the development of more effective flavonoid analogues. UCCF-029 Free base demonstrates improved potency compared to apigenin in activating wild-type CFTR. UCCF-029 Free base also exhibits potential for activating the mutant CFTR (G551D-CFTR) though not as robustly as apigenin.
|
-

- HY-E70765
-
|
|
Raf
|
Cancer
|
|
Ras-associated factor -1 (RAF1) belongs to the RAF protein kinases family, also known as C-Raf. RAF1 participates in Ras-RAF-MEK-ERk signaling pathway (MAPK signaling pathway), and transmits extracellular signals into the nucleus through cell membrane receptors, thereby mediating the expression of intracellular specific proteins and participating in the regulation of cell proliferation, differentiation, apoptosis, autophagy and other functions. RAF1 YDYD is a mutant of RAF1. RAF1 YDYD Recombinant Human Active Protein Kinase is a recombinant RAF1 YDYD protein that can be used to study RAF1 YDYD-related functions .
|
-

- HY-171958
-
|
|
PROTACs
EGFR
|
Cancer
|
|
PROTAC EGFR degrader 12 (example 1) is a PROTAC degrader targeting EGFR that can effectively degrade EGFR mutants, but has little effect on EGFR WT. PROTAC EGFR degrader 12 shows IC50s against EGFR L858R-T790M (NCI-H1975 cells), EGFR L858R (NCI-H3255 cells), and EGFR L858R-T790M-L797S (NCI-H1975+CS cells) of all <50 nM .
|
-

- HY-E70757
-
|
|
c-Met/HGFR
|
Cancer
|
|
Mesenchymal-to-epithelial transition (MET) is a receptor tyrosine kinase for hepatocyte growth factor (HGF). MET overactivation is strongly associated with angiogenesis, cellular motility, growth, and invasion. Aberrant MET signaling can drive tumorigenesis in several cancer types through various molecular mechanisms, including MET amplification, MET exon 14 skipping mutation, MET overexpression, and MET fusions. MET Y1230H is a mutant of MET. MET Y1230H Recombinant Human Active Protein Kinase is a recombinant MET Y1230H protein that can be used to study MET Y1230H-related functions .
|
-

- HY-111373
-
|
|
mTOR
Autophagy
|
Cancer
|
|
RapaLink-1, the third-generation bivalent mTOR inhibitor, combines Rapamycin (HY-10219) with MLN0128 (HY-13328, a second-generation mTOR kinase inhibitor) by an inert chemical linker. RapaLink-1 shows better efficacy than Rapamycin or mTOR kinase inhibitors (TORKi), potently blocking cancer-derived, activating mutants of mTOR. RapaLink-1 can cross the blood-brain barrier. RapaLink-1 binding to FKBP12 results in targeted and durable inhibition of mTORC1. RapaLink-1 plays an antithrombotic role in antiphospholipid syndrome by improving autophagy. Anticancer activity .
|
-

- HY-E70751
-
|
|
c-Met/HGFR
|
Cancer
|
|
Mesenchymal-to-epithelial transition (MET) is a receptor tyrosine kinase for hepatocyte growth factor (HGF). MET overactivation is strongly associated with angiogenesis, cellular motility, growth, and invasion. Aberrant MET signaling can drive tumorigenesis in several cancer types through various molecular mechanisms, including MET amplification, MET exon 14 skipping mutation, MET overexpression, and MET fusions. MET G1163R is a mutant of MET. MET G1163R Recombinant Human Active Protein Kinase is a recombinant MET G1163R protein that can be used to study MET G1163R-related functions .
|
-

- HY-152233
-
|
|
HIV
|
Infection
|
|
Reverse transcriptase-IN-4 (compound F10) is a potent and selective non-nucleoside reverse transcriptase (NNRT) inhibitor with an EC50 value of 0.053 μM for wild-type HIV-1 and an EC50 value of 0.26 μM for HIV-1 mutant E138K . Reverse transcriptase-IN-4 is a click chemistry reagent, it contains an Azide group and can undergo copper-catalyzed azide-alkyne cycloaddition reaction (CuAAc) with molecules containing Alkyne groups. It can also undergo strain-promoted alkyne-azide cycloaddition (SPAAC) reactions with molecules containing DBCO or BCN groups.
|
-

- HY-17499R
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
EGFR-IN-12 (Standard) is the analytical standard of EGFR-IN-12. This product is intended for research and analytical applications. EGFR-IN-12 is a 4,6-disubstituted pyrimidine and is a potent, ATP-competitive, irreversible and highly selective EGFR inhibitor with an IC50of 21 nM. EGFR-IN-12 also inhibits mutant EGFRL858R and EGFRL861Q with IC50s of 63 nM and 4 nM, respectively. EGFR-IN-12 displays strong selectivity for EGFR over HER4 (IC50 = 7640 nM) and a panel of 55 other kinases. EGFR-IN-12 induces cells apoptosis and has antitumor activity .
|
-

- HY-P991571
-
|
GC-1118A
|
EGFR
PERK
Akt
|
Cancer
|
|
GC1118 (GC-1118A) is a fully human anti-EGFR monoclonal antibody with binding affinity of 0.16 nM (KD) to EGFR. GC1118 displays potent inhibitory effects on high- and low-affinity EGFR ligand-induced signaling. GC1118 shows potent anti proliferative activity in KRAS wild-type and KRAS mutant cells. GC1118 can reach the tumor by crossing both BBB (blood-brain barrier) and BTB (brain-tumor barrier) and shows superior anti-tumor effects in various mice xenograft models. GC1118 can be used for the researches of cancer, such as colorectal cancer .
|
-

- HY-148409
-
|
|
Ferroptosis
Apoptosis
Autophagy
MDM-2/p53
|
Cancer
|
|
MMRi62, a ferroptosis inducer targeting MDM2-MDM4 (negative regulators of tumor suppressor p53). MMRi62 shows a P53-independent pro-apoptotic activity against pancreatic ductal adenocarcinoma (PDAC) cells and induce autophagy. MMRi62 inducesferroptosis, resulting in a increase of reactive oxygen and lysosomal degradation of ferritin heavy chain (FTH1). MMRi62 also leads to proteasomal degradation of mutant p53, also inhibits orthotopic xenograft PDAC mouse model in vivo with high frequency mutation characteristics of KRAS and TP53.12 .
|
-

- HY-E70753
-
|
|
c-Met/HGFR
|
Cancer
|
|
Mesenchymal-to-epithelial transition (MET) is a receptor tyrosine kinase for hepatocyte growth factor (HGF). MET overactivation is strongly associated with angiogenesis, cellular motility, growth, and invasion. Aberrant MET signaling can drive tumorigenesis in several cancer types through various molecular mechanisms, including MET amplification, MET exon 14 skipping mutation, MET overexpression, and MET fusions. MET M1250T is a mutant of MET. MET M1250T Recombinant Human Active Protein Kinase is a recombinant MET M1250T protein that can be used to study MET M1250T-related functions .
|
-

- HY-157288
-
|
|
Biochemical Assay Reagents
|
Metabolic Disease
|
|
(R)-Phe-A110/B319, a hapten, is a selective binder to tumor-associated antigens. (R)-Phe-A110/B319 has a 20-fold higher affinity towards the H1047R mutant of p110α in the p110α/p85α PI3K complex. (R)-Phe-A110/B319 can be used for the research of conditional chimeric antigen receptor T (CAR-T) cell activation and tumor targeting .
|
-

- HY-E70750
-
|
|
c-Met/HGFR
|
Cancer
|
|
Mesenchymal-to-epithelial transition (MET) is a receptor tyrosine kinase for hepatocyte growth factor (HGF). MET overactivation is strongly associated with angiogenesis, cellular motility, growth, and invasion. Aberrant MET signaling can drive tumorigenesis in several cancer types through various molecular mechanisms, including MET amplification, MET exon 14 skipping mutation, MET overexpression, and MET fusions. MET F1200I is a mutant of MET. MET F1200I Recombinant Human Active Protein Kinase is a recombinant MET F1200I protein that can be used to study MET F1200I-related functions .
|
-

- HY-170665
-
|
|
EGFR
|
Cancer
|
|
EGFR T790M/L858R-IN-9 (Compound 8) is an EGFR-L858R/T790M inhibitor that demonstrates potent inhibitory phosphorylation effects against the EGFR-L858R/T790M mutant kinase, with an IC50 value of 0.0064µM. EGFR T790M/L858R-IN-9 also inhibits the proliferation of non-small cell lung cancer (NSCLC) cells and can be utilized in cancer research .
|
-

- HY-116762
-
|
|
Biochemical Assay Reagents
|
Others
|
|
Quorum sensing is a regulatory system used by bacteria to control gene expression in response to increased cell density. The control of bacterial infection by quenching the quorum sensing system of bacteria is a promising research area. The expression of specific target genes, such as transcriptional regulators belonging to the LuxIR protein family, is coordinated by the synthesis of diffusible acyl homoserine lactone (AHL) molecules. N-butyryl-L-Homocysteine thio-lactone is an analog of N-butyryl-L-homoserine lactone, a small, diffusible signaling molecule involved in quorum sensing, thereby controlling gene expression and cellular metabolism . N-butyryl-L-homocysteine thiolactone induces violacein expression in Viola viola mutants that normally fail to produce AHL.
|
-

- HY-158684
-
|
|
PROTACs
MDM-2/p53
|
Cancer
|
|
YX-02-030M is a PROTAC MDM2 degrader. YX-02-030M inhibits MDM2-p53 binding and VHL-HIF1α binding with IC50s of 63 nM and 1.35 μM respectively. YX-02-030M binds MDM2 and recruits the VHL E3 ubiquitin ligase to initiate MDM2 degradation, and effectively kills p53 mutant or deleted Triple-negative breast cancers (TNBC) cells. (Blue: VHL ligand; Black: linker; Pink: MDM2 inhibitor) .
|
-

- HY-E70711
-
|
|
EGFR
|
Cancer
|
|
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR T790M/L858R Recombinant Human Active Protein Kinase is a recombinant EGFR T790M/L858R protein that can be used to study EGFR T790M/L858R-related functions .
|
-

- HY-13238S2
-
|
S/GSK1349572-d5
|
Isotope-Labeled Compounds
HIV Integrase
HIV
|
Infection
|
|
Dolutegravir-d5 is deuterium labeled Dolutegravir. Dolutegravir (S/GSK1349572) is a highly potent and orally bioavailable HIV integrase strand transfer inhibitor with an IC50 of 2.7 nM for HIV-1 integrase-catalyzed strand transfer. Dolutegravir (S/GSK1349572) inhibits HIV-1 viral replication with an IC50 of 0.51 nM in peripheral blood mononuclear cells. Dolutegravir retains a high potency against the HIV-1 Y143R, N155H, and G140S/Q148H mutants (EC50=3.6-5.8 nM) .
|
-

- HY-13238A
-
|
S/GSK1349572 sodium
|
HIV Integrase
HIV
|
Infection
|
|
Dolutegravir sodium (S/GSK1349572 sodium) is a highly potent and orally bioavailable HIV integrase strand transfer inhibitor with an IC50 of 2.7 nM for HIV-1 integrase-catalyzed strand transfer. Dolutegravir sodium (S/GSK1349572 sodium) inhibits HIV-1 viral replication with an IC50 of 0.51 nM in peripheral blood mononuclear cells. Dolutegravir sodium (S/GSK1349572 sodium) retains a high potency against the HIV-1 Y143R, N155H, and G140S/Q148H mutants (EC50=3.6-5.8 nM) .
|
-

- HY-131864A
-
|
|
PROTACs
EGFR
|
Cancer
|
|
SJF-1528 (PROTAC 1) hemihydrate is a potent EGFR PROTAC degrader with DC50 values of 39.2 nM and 736.2 nM for wild-type EGFR in OVCAR8 cells and Exon 20 Ins mutant EGFR in HeLa cells. SJF-1528 hemihydrate also degrades HER2. SJF-1528 hemihydrate promotes ubiquitination and degradation of EGFR and can be used in breast cancer research (Pink: ligand for target protein (HY-159047); Black: linker Tos-PEG2-CH2-Boc (HY-130532); Blue: ligand for E3 ligase (S,R,S)-AHPC (HY-125845)) .
|
-

- HY-N2099A
-
|
(Z)-Senegin III
|
Drug Derivative
|
Neurological Disease
Metabolic Disease
Inflammation/Immunology
|
|
(Z)-Onjisaponin B ((Z)-Senegin III) is a derivative of Onjisaponin B (HY-N2099). Onjisaponin B is an orally active natural product derived from Polygala tenuifolia. Onjisaponin B inhibits NF-κB p65. Onjisaponin B enhances autophagy and accelerates the degradation of mutant α-synuclein and huntingtin. Onjisaponin B reduces β-amyloid (Aβ) production. Onjisaponin B reduces radiation-induced cell apoptosis. Onjisaponin B has anti-oxidant and anti-inflammatory activities. Onjisaponin B can be used for neurological disease and radiation injury study, and its metabolite tenuifolin (TF) can enter the brain through the BBB .
|
-

- HY-117807
-
|
|
Ras
Farnesyl Transferase
VEGFR
|
Cancer
|
|
A-176120 is a selective inhibitor of farnesyl transferase (IC50=1.2 nM) based on a farnesyl pyrophosphate (FPP) analog, with superior selectivity against GGTaseI (IC50=423 nM), GGTaseII (IC50=3000 nM), and SSase (IC50>10 μM). A-176120 inhibits ras processing in H-ras transformed NIH3T3 cells and HCT116 K-ras mutant cells (ED50=1.6 and 0.5 μM, respectively). A-176120 has antiangiogenic and antitumor activities in vivo and reduces capillary structure formation and VEGF secretion .
|
-

- HY-E70701
-
|
|
EGFR
|
Cancer
|
|
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR d747-749/A750P Recombinant Human Active Protein Kinase is a recombinant EGFR d747-749/A750P protein that can be used to study EGFR d747-749/A750P-related functions .
|
-

- HY-178059
-
|
|
FGFR
p38 MAPK
Akt
ERK
Apoptosis
|
Cancer
|
|
FGFR-IN-23 (Compound 9p) is a covalent pan FGFR inhibitor with IC50s of 14, 4.2, 5 and 220 nM for FGFR1, FGFR2, FGFR3 and FGFR4, respectively. FGFR-IN-23 also has potent inhibitory activity against gatekeeper mutants, such as FGFR1 V561M and FGFR3 V555M. FGFR-IN-23 suppresses the activation of FGFR-mediated signaling and induces apoptosis. FGFR-IN-23 shows significant antitumor efficacy in RT112 xenograft mouse models. FGFR-IN-23 can be used for cancers and its drug resistance research .
|
-

- HY-126247B
-
|
|
Drug Derivative
Ras
|
Cancer
|
|
(R)-BI-2852 is the isomer of BI-2852 (HY-126247), and can be used as an experimental control. BI-2852 is a KRAS inhibitor for the switch I/II pocket (SI/II-pocket) by structure-based agent design with nanomolar affinity. BI-2852 is mechanistically distinct from covalent KRASG12C inhibitor (binds to switch II pocket) and binds ten-fold more strongly to active KRASG12D versus KRASwt (740 nM vs 7.5 μM). BI-2852 blocks GEF, GAP, and effector interactions with KRAS, leading to inhibition of downstream signaling and an antiproliferative effect in KRAS mutant cells.
|
-

- HY-E70842
-
|
|
MEK
|
Cancer
|
|
MAP2K1 (also known as MEK1) is downstream of the RAF family and activation results in ERK1/2 activation. Activating mutations in MAP2K1 have been reported almost exclusively in exons 1 and 2 in both hematologic malignancies. MAP2K1 L115P is a mutant of MAP2K1. MAP2K1 L115P Recombinant Human Active Protein Kinase is a recombinant MAP2K1 L115P protein that can be used to study MAP2K1 L115P-related functions .
|
-

- HY-N16410
-
|
5-cis-C12-HSL
|
Bacterial
|
Infection
|
|
N-cis-Dodec-5(Z)-enoyl-L-homoserine lactone (5-cis-C12-HSL) (Compound 2) is an acylated homoserine lactone. N-cis-Dodec-5(Z)-enoyl-L-homoserine lactone can be isolated for Mesorhizobium sp. N-cis-Dodec-5(Z)-enoyl-L-homoserine lactone restores protease and pyoverdin production of an AHL-deficient Pseudomonas aeruginosa PAO1 lasI rhlI double mutant. N-cis-Dodec-5(Z)-enoyl-L-homoserine lactone has no significant antibacterial activity and cytotoxicity against tumor cells .
|
-

- HY-175326
-
|
|
SOS1
|
Cancer
|
|
SOS1-IN-21 is an orally active inhibitor of son of Sevenless 1 (SOS1) with an IC50 of 15 nM. SOS1 is a guanine nucleotide exchange factor (GEF) that activates KRAS by facilitating the exchange of GDP for GTP. SOS1-IN-21 exhibits potent antiproliferative activity, with IC50 values of 16 nM in NCI-H358 and 17 nM in Mia Paca-2 cell proliferation assays. SOS1-IN-21 exhibits significant antitumor activity in the Mia Paca-2 xenograft model. SOS1-IN-21 can be used for the study of KRAS mutant tumors, such as pancreatic cancer .
|
-

- HY-13238S1
-
|
S/GSK1349572-d3
|
Isotope-Labeled Compounds
HIV Integrase
HIV
|
Infection
|
|
Dolutegravir-d3 is the deuterium labeled Dolutegravir. Dolutegravir (S/GSK1349572) is a highly potent and orally bioavailable HIV integrase strand transfer inhibitor with an IC50 of 2.7 nM for HIV-1 integrase-catalyzed strand transfer. Dolutegravir (S/GSK1349572) inhibits HIV-1 viral replication with an IC50 of 0.51 nM in peripheral blood mononuclear cells. Dolutegravir retains a high potency against the HIV-1 Y143R, N155H, and G140S/Q148H mutants (EC50=3.6-5.8 nM) .
|
-

- HY-131864
-
|
|
PROTACs
EGFR
|
Cancer
|
|
SJF-1528 (PROTAC 1) is a potent EGFR PROTAC degrader with DC50 values of 39.2 nM and 736.2 nM for wild-type EGFR in OVCAR8 cells and Exon 20 Ins mutant EGFR in HeLa cells. SJF-1528 also degrades HER2. SJF-1528 promotes ubiquitination and degradation of EGFR and can be used in breast cancer research (Pink: ligand for target protein (HY-159047); Black: linker Tos-PEG2-CH2-Boc (HY-130532); Blue: ligand for E3 ligase (S,R,S)-AHPC (HY-125845)) .
|
-

- HY-W745090
-
|
|
Formyl Peptide Receptor (FPR)
Src
ERK
Akt
p38 MAPK
|
Others
|
|
Isomaltulose monohydrate is a fMLP inhibitor and also inhibits Src kinase, ERK1/2, p38 and AKT phosphorylation signals in immune regulation. Isomaltulose monohydrate can interfere with the interaction between the βγ subunit of the fMLP receptor Gi protein and its downstream molecules, thereby inhibiting fMLP-induced respiratory burst. Isomaltulose monohydrate inhibits fMLP (0.1 μM)-induced superoxide anion production (IC50: 1.98 μM) , cathepsin G release (IC< sub>50: 2.76 μM) and chemotaxis. Isomaltulose monohydrate can improve excessive activation of neutrophils and reduce inflammation or tissue damage. A series of derivatives of Isomaltulose monohydrate are found to have inhibitory effects on FSGS-related TRPC6 functional mutants .
|
-

- HY-E70696
-
|
|
EGFR
|
Cancer
|
|
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR C797S/L858R Recombinant Human Active Protein Kinase is a recombinant EGFR C797S/L858R protein that can be used to study EGFR C797S/L858R-related functions .
|
-

- HY-E70698
-
|
|
EGFR
|
Cancer
|
|
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR d746-750/C797S Recombinant Human Active Protein Kinase is a recombinant EGFR d746-750/C797S protein that can be used to study EGFR d746-750/C797S-related functions .
|
-

- HY-19932
-
|
|
HCV Protease
HCV
|
Infection
|
|
MK-2748 is a hepatitis C virus (HCV) NS3/4a protease inhibitor. MK-2748 exhibits broad-spectrum and potent antiviral activity, with nanomolar-level activity against all genotypes (gt1a-gt6) (IC₅₀ < 0.115 nM), showing only slightly weaker activity against gt3a (1.212 nM), and maintaining high inhibitory activity against drug-resistant mutant strains such as gt1b R155K (IC₅₀ = 0.032 nM) and D168Y (IC₅₀ = 0.057 nM). MK-2748 can be used in the research of HCV infection .
|
-

- HY-159958
-
|
|
Potassium Channel
|
Neurological Disease
|
|
KNa1.1-IN-2 (Compound Z05) is a selective KNa1.1 channel inhibitor with blood-brain barrier penetration, particularly effective against the hERG channel. KNa1.1-IN-2 works by binding to the KNa1.1 channel and blocking the channel activity induced by Gain-of-function (GOF) mutations, effectively intervening in KCNT1-related epilepsy. Additionally, KNa1.1-IN-2 inhibits the GOF mutant Y796H. KNa1.1-IN-2 holds promise for research into KCNT1-related epilepsy disorders .
|
-

- HY-112289B
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
(1R)-IDH889 is the isomer of IDH889 (HY-112289), and can be used as an experimental control. IDH889 is an orally available, brain penetrant, allosteric and mutant specific inhibitor of isocitrate dehydrogenase 1 (IDH1). IDH889 has potent selectivity for IDH1 R132* mutations, with IC50s of 0.02 μM, 0.072 μM and 1.38 μM for IDH1 R132H, IDH1 R132C and IDH1 wt, respectively. IDH889 shows potent cellular inhibition of R-2-hydroxyglutarate (2-HG) production with an IC50 of 0.014 μM .
|
-

- HY-175804
-
|
|
SARS-CoV
|
Infection
|
|
M2 ion channel blocker-2 (Compound 10) is a M2 channel blocker. M2 ion channel blocker-2 significantly blocks wild-type and mutant M2 (L27F and V27A) ion channels. M2 ion channel blocker-2 has potent antiviral activity against HCoV-229E (EC50: 4.7 μM in cytopathic effect) but not against influenza A virus. M2 ion channel blocker-2 has no significant inhibition of hERG and cytochrome P450 (CYP1A2, CYP2C19, and CYP3A4) activity .
|
-

- HY-403733A
-
|
|
Androgen Receptor
|
Cancer
|
|
(-)-JJ-450 is a non-competitive antagonist targeting the androgen receptor (AR). (-)-JJ-450 is more potent than (+)-JJ-450 (HY-403733B) in inhibiting androgen-induced AR activity, and the mechanism of AR inhibition by (+)-JJ-450 is different from that of Enzalutamide (MDV3100) (HY-70003), which may target the ligand binding domain (LBD) of AR. (-)-JJ-450 inhibits the transcriptional activity of wild-type AR and mutant AR F876L by inhibiting AR nuclear translocation and promoting nuclear degradation of unbound AR. (-)-JJ-450 can be used in the study of castration-resistant prostate cancer (CRPC) resistant to Enzalutamide .
|
-

- HY-168012
-
|
|
Ras
Phosphatase
|
Cancer
|
|
Pan-RAS-IN-6 (compound 24) is an inhibitor targeting DUSP6, which reduces MAPK activation in the brain of the NCI-H1373-Luc model (DUSP6), at the same time, it shows significant tumor growth inhibition and tumor regression effects in the NSCLC brain metastasis mouse model. Pan-RAS-IN-6 shows high selectivity and strong inhibitory effects, especially in KRAS mutation-related signaling pathways, demonstrating varying inhibitory activity against different KRAS mutants and interacting proteins. The IC50 values for KRAS G12C, G12D, and G12V are 1.3 nM, 4.7 nM, and 0.3 nM, respectively .
|
-

- HY-149508
-
|
|
Keap1-Nrf2
Reactive Oxygen Species (ROS)
|
Cancer
|
|
Nrf2-IN-3 (Compound R16) is a small-molecule NRF2 inhibitor and increases reactive oxygen species (ROS) production. Nrf2-IN-3 selectively binds KEAP1 mutants and restores their NRF2-inhibitory function by repairing the disrupted KEAP1/NRF2 interactions, leading to proteasome-dependent NRF2 degradation in cells. Nrf2-IN-3 sensitizes KEAP1-mutated tumor cells to Cisplatin (HY-17394), Gefitinib (HY-50895), and KEAP1 G333C-mutated xenograft to Cisplatin .
|
-

- HY-128888B
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
(S,R)-GSK321 is the (S,R)-enantiomer of GSK321 (HY-18948). GSK321 is a potent, selective mutant IDH1 inhibitor with IC50 values of 2.9, 3.8, 4.6 and 46 nM for R132G, R132C, R132H and WT IDH1, respectively, and >100-fold selectivity over IDH2. GSK321 induces decrease in intracellular 2-HG, abrogation of the myeloid differentiation block and induction of granulocytic differentiation at the level of leukemic blasts and more immature stem-like cells. GSK321can be used for research of acute myeloid leukemia (AML) and other cancers .
|
-

- HY-161785
-
|
|
Apoptosis
EGFR
|
Cancer
|
|
EGFR-IN-117 (Compound 8h) exhibits inhibitory activity against EGFR mutation, targets the tumor environment, and induces apoptosis of cancer cells. EGFR-IN-117 inhibits proliferations of H1975, PC-9, and EGFR mutant cells BaF3-EGFR L858R/T790M/C797S and BaF3– C797S/Del19/T790M, with IC50 of 13 nM, 19 nM, 1.2 nM and 1.3 nM, respectively. EGFR-IN-117 exhibits antitumor efficacy in mouse models .
|
-

- HY-139920
-
|
SH-1028
|
EGFR
|
Cancer
|
|
Oritinib (SH-1028), an irreversible third-generation EGFR TKI, overcomes T790M-mediated resistance in non-small cell lung cancer. Oritinib (SH-1028), a mutant-selective inhibitor of EGFR kinase activity, inhibits EGFR WT, EGFR L858R, EGFR L861Q, EGFR L858R/T790M, EGFR d746-750 and EGFR d746-750/T790M kinases, with IC50s of 18, 0.7, 4, 0.1, 1.4 and 0.89 nM, respectively .
|
-

- HY-116572
-
|
|
Reactive Oxygen Species (ROS)
JNK
Apoptosis
Caspase
|
Cancer
|
|
TASIN-1 hydrochloride is a selective inhibitor of truncated APC TR (adenomatous polyposis coli gene) that exerts cytotoxic effects by inhibiting cholesterol biosynthesis. TASIN-1 hydrochloride specifically targets colorectal cancer (CRC) cells carrying APC truncated mutations, while having no significant toxicity to wild-type APC cells. TASIN-1 hydrochloride exerts cytotoxic effects by targeting Emopamil binding protein (EBP) to inhibit cholesterol biosynthesis, triggering endoplasmic reticulum (ER) stress, reactive oxygen species (ROS) generation, and JNK-mediated apoptosis, and inhibiting Akt survival signaling. TASIN-1 hydrochloride can be used to prevent and intervene in APC mutant colorectal cancer .
|
-

- HY-E70702
-
|
|
EGFR
|
Cancer
|
|
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR d747-752/P753S Recombinant Human Active Protein Kinase is a recombinant EGFR d747-752/P753S protein that can be used to study EGFR d747-752/P753S-related functions .
|
-

- HY-W744699
-
|
(+)-Larixol
|
Src
ERK
Akt
|
Inflammation/Immunology
|
|
Larixol is an fMLP inhibitor and also inhibits Src kinase, ERK1/2, p38 and AKT phosphorylation signals in immune regulation. Larixol can interfere with the interaction between the βγ subunit of the fMLP receptor Gi protein and its downstream molecules, thereby inhibiting fMLP-induced respiratory burst. Larixol inhibits fMLP (0.1 μM)-induced superoxide anion production (IC50: 1.98 μM), cathepsin G release (IC50: 2.76 μM), and chemotaxis. Larixol improves neutrophil hyperactivation and reduces inflammation or tissue damage. A series of Larixol derivatives were found to have inhibitory effects on FSGS-related TRPC6 functional mutants .
|
-

- HY-175541
-
|
|
PROTACs
Btk
|
Cancer
|
|
TQ-3959 is an orally active BTK PROTAC degrader, with a DC50 of 14.6 nM. TQ-3959 exerts antiproliferative activity against both wild-type BTK and C481S mutant BTK cell lines. TQ-3959 exhibits tumor growth inhibition in female NOD-SCID mice bearing TMD-8 xenografts. TQ-3959 can be used in the research on B-cell malignancies such as lymphoma.(Pink: BTK ligand (HY-150885), Blue: CRBN Ligand (HY-W733888), Black: Linker (HY-W061884), E3 ligase ligand-linker conjugate (HY-175545)) .
|
-

- HY-116572A
-
|
|
Reactive Oxygen Species (ROS)
JNK
Apoptosis
Caspase
|
Cancer
|
|
TASIN-1 is a selective inhibitor of truncated APC TR (adenomatous polyposis coli gene) that exerts cytotoxic effects by inhibiting cholesterol biosynthesis. TASIN-1 specifically targets colorectal cancer (CRC) cells carrying APC truncated mutations, while having no significant toxicity to wild-type APC cells. TASIN-1 exerts cytotoxic effects by targeting Emopamil binding protein (EBP) to inhibit cholesterol biosynthesis, triggering endoplasmic reticulum (ER) stress, reactive oxygen species (ROS) generation, and JNK-mediated apoptosis, and inhibiting Akt survival signaling. TASIN-1 can be used to prevent and intervene in APC mutant colorectal cancer .
|
-

- HY-173054
-
|
|
Bacterial
|
Infection
|
|
FtsZ-IN-12 (Compound 16e) is the inhibitor for filamentous temperature-sensitive mutant Z (FtsZ) that promotes the polymerization of FtsZ protein, inhibits its GTPase activity, thereby interfering with bacterial cell division process. FtsZ-IN-12 exhibits boardspectrum antibacterial activity that inhibits B. subtilis ATCC9372, B. pumilus CMCC63202, S. aureus ATCC25923, E. coli BW25113 and A. baumannii ATCC19606 with MIC of 0.062-1 μg/mL. FtsZ-IN-12 inhibits the formation of bacterial biofilms and exhibits a clearing effect on mature biofilms. FtsZ-IN-12 exhibits bactericidal activity without hemolytic toxicity to mammalian red blood cells (15 mg/kg) .
|
-

- HY-B0984R
-
|
|
Calcium Channel
Ras
STING
Autophagy
|
Infection
Cardiovascular Disease
Cancer
|
|
Fendiline (hydrochloride) (Standard) is the analytical standard of Fendiline (hydrochloride). This product is intended for research and analytical applications. Fendiline hydrochloride, a diphenylalkylamine type of antianginal agent, is an L-type calcium channel blocker (IC50 of 17 µM). Fendiline hydrochloride is also a selective K-Ras inhibitor, and has no effect on H-Ras and N-Ras. Fendiline hydrochloride inhibits K-Ras plasma membrane localization (IC50 of 9.64 μM), inhibits K-Ras signal output and blocks the proliferation of pancreatic, colon, lung, and endometrial cancer cell lines expressing oncogenic mutant K-Ras. Fendiline hydrochloride is a STING agonist and is able to inhibit the growth of multiple refractory cold tumors (MC38, CT26 and B16F10) .
|
-

- HY-13238R
-
|
S/GSK1349572 (Standard)
|
Reference Standards
HIV Integrase
HIV
|
Infection
|
|
Dolutegravir (Standard) is the analytical standard of Dolutegravir. This product is intended for research and analytical applications. Dolutegravir (S/GSK1349572) is a highly potent and orally bioavailable HIV integrase strand transfer inhibitor with an IC50 of 2.7 nM for HIV-1 integrase-catalyzed strand transfer. Dolutegravir (S/GSK1349572) inhibits HIV-1 viral replication with an IC50 of 0.51 nM in peripheral blood mononuclear cells. Dolutegravir retains a high potency against the HIV-1 Y143R, N155H, and G140S/Q148H mutants (EC50=3.6-5.8 nM) .
|
-

- HY-175344
-
|
|
SARS-CoV
Virus Protease
Ser/Thr Protease
|
Infection
Inflammation/Immunology
|
|
TMP1 is an orally active bispecific inhibitor of M pro (IC50 = 312.5 nM)/TMPRSS2 (IC50 = 1.28 μM, KD = 10.10 μM). TMP1 exhibits broad protection against different SARS-CoV-2 variants in vitro. TMP1 cross-protects against highly pathogenic coronaviruses (SARS-CoV-1, SARS-CoV-2, and MERS-CoV) in vivo and effectively blocks the transmission of SARS-CoV-2. TMP1 can inhibit infection by SARS-CoV-2 escape mutants that are resistant to Nivolumab (HY-P9903). TMP1 can be used in coronavirus research .
|
-

- HY-113137
-
|
|
Endogenous Metabolite
|
Cancer
|
|
N2,N2-Dimethylguanosine is a methylated nucleoside present in RNA and a structural modification component of tRNA. N2,N2-Dimethylguanosine hinders reverse transcriptase-mediated cDNA synthesis and is one of the key modifications that affect sequencing efficiency in high-throughput RNA sequencing. N2,N2-Dimethylguanosine can selectively remove one of the methyl groups through AlkB mutant enzymes (such as D135S/L118V) and convert it into N2-methylguanosine, thereby reducing the hindrance to reverse transcription .
|
-

- HY-136265
-
|
|
Aminoacyl-tRNA Synthetase
|
Cancer
|
|
BC-LI-0186 is a potent and selective inhibitor of Leucyl-tRNA synthetase (LRS; LeuRS) and Ras-related GTP-binding protein D (RagD) interaction (IC50=46.11 nM). BC-LI-0186 competitively binds to the RagD interacting site of LRS (Kd=42.1 nM) and has on effects on LRS-Vps34, LRS-EPRS, RagB-RagD association, mTORC1 complex formation or the activities of 12 kinases. BC-LI-0186 can effectively suppress the activity of cancer-associated?MTOR?mutants and the growth of rapamycin-resistant cancer cells.?BC-LI-0186 is a promising agent for lung cancer research .
|
-

- HY-157228
-
|
|
PROTACs
Ras
|
Cancer
|
|
ACBI3 (compound 7), a chemical probe, is a PROTAC targeting KRAS. ACBI3 is composed of PROTAC target protein ligand pan-KRAS degrader 1 (HY-162960) (red part), E3 ligase ligand E3 ligase Ligand 43 (HY-401613) (blue part) and PROTAC Linker 1-Bromo-4-(ethynyloxy)butane (HY-169992) (black part), among which the conjugate of E3 ubiquitin ligase ligand + Linker is E3 Ligase Ligand-linker Conjugate 143 (HY-169995). ACBI3 achieves in vivo degradation of oncogenic KRAS, resulting in durable pathway modulation and tumor regressions in KRAS mutant xenograft mouse models .
|
-

- HY-156086
-
|
|
Trk Receptor
|
Cancer
|
|
TRK-IN-24 (compound 10g) is a Trk Receptor inhibitor that inhibits TRKA, TRKC, TRKA G595R, TRKA G667C and TRKA F589L IC50s are 5.21, 4.51, 6.77, 1.42 and 6.13 nM respectively. TRK-IN-24 has antitumor efficacy in BaF3-CD74-NTRK1 G595R and BaF3-CD74-NTRK1 G667C xenograft models. TRK-IN-24 inhibits the proliferation of Ba/F3 cells transfected with single mutants such as SF, GK, and xDFG, with an IC50 of 1.43-47.56 nM .
|
-

- HY-122562
-
MT-802
3 Publications Verification
|
PROTACs
Btk
|
Cancer
|
|
MT-802 is a BTK PROTAC degrader. MT-802 degrades wild-type BTK (DC50 = 14.6 nM) and BTK mutants including E41K, C481S (DC50 = 14.9 nM), C481R, C481Y, C481T, C481F, L528W, and inhibits their Y223 phosphorylation. BI-4732 can be used for the study of Ibrutinib (HY-10997)-resistant chronic lymphocytic leukemia (CLL). (Pink: BTK ligand (HY-150885), Blue: CRBN Ligand (HY-14658), Black: Linker (HY-141371), E3 ligase ligand-linker conjugate (HY-176340)) .
|
-

- HY-E70740
-
|
|
c-Kit
|
Cancer
|
|
KIT (CD117) is an important cell surface marker used to identify certain types of hematopoietic(blood) progenitors in the bone marrow. KIT is a cytokine receptor expressed on the surface of hematopoietic stem cells as well as other cell types. Altered forms of this receptor may be associated with some types of cancer. KIT V559D/V654A is a mutant of KIT. KIT V559D/V654A Recombinant Human Active Protein Kinase is a recombinant KIT V559D/V654A protein that can be used to study KIT V559D/V654A-related functions .
|
-

- HY-15800
-
|
|
TNK1
LRRK2
|
Neurological Disease
Inflammation/Immunology
|
|
CZC-25146 hydrochloride is a potent LRRK2 inhibitor with IC50 values of 4.76 nM and 6.87 nM for wild-type LRRK2 and G2019S LRRK2, respectively. CZC-25146 hydrochloride inhibits PLK4, GAK, TNK1, CAMKK2 and PIP4K2C as well. CZC-25146 hydrochloride prevents mutant LRRK2-induced injury of neurons in vitro. CZC-25146 hydrochloride exhibits relatively favorable pharmacokinetic properties in mice. CZC-25146 hydrochloride can increase normal α-1-antitrypsin (AAT) secretion and reduce inflammatory cytokines. CZC-25146 hydrochloride can be used to research Parkinson's disease and liver diseases .
|
-

- HY-D0098
-
|
N-(5-Fluoresceinyl)maleimide
|
Fluorescent Dye
|
Others
|
Fluorescein-5-maleimide (N-(5-Fluoresceinyl)maleimide) is a fluorescent dye. Fluorescein-5-maleimide can be used to detect the redox state of thiols in eukaryotic cells. Fluorescein-5-maleimide can label peptides and is used to detect negatively charged nanoparticles. Fluorescein-5-maleimide can also label actin to explore its interaction with cardiac myosin-binding protein C (cMyBP-C), which helps in developing small molecule modulators for heart failure. Fluorescein-5-maleimide can screen mutant proteins that contain cysteine residues. The excitation wavelength of Fluorescein-5-maleimide is 494 nm, and the emission wavelength is 519 nm .
|
-

- HY-175302
-
|
|
Trk Receptor
Apoptosis
|
Cancer
|
|
TRK-IN-32 is a potent TRK inhibitor. TRK-IN-32 potently inhibits TRK WT, TRK G595R and TRK G667C with IC50 values of 0.08 nM, 2.14 nM and 0.68 nM, respectively. TRK-IN-32 also demonstrates antiproliferative activity against a panel of Ba/F3 cell lines transformed with wild type, xDFG, solvent-front as well as gatekeeper mutant TRK fusion proteins. TRK-IN-32 induces apoptosis of Ba/F3-TRKA WT and Ba/F3-TRKA G667C cells.TRK-IN-32 can be used for the study of various cancers (such as thyroid cancer, secretory breast carcinoma) .
|
-

- HY-104042
-
|
AG-881
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
Vorasidenib (AG-881) is an orally available, brain penetrant second-generation dual mutant isocitrate dehydrogenases 1 and 2 (mIDH1/2) inhibitor. Vorasidenib (AG-881) exhibits nanomolar inhibition of (D)-2-hydroxyglutarate (D-2-HG), and the IC50 ranges of 0.04~22 nM against IDH1 R132C, IDH1 R132G, IDH1 R132H and IDH1 R132S and 7~14 nM against IDH2 R140Q and 130 nM against IDH2 R172K. Vorasidenib can be used for the study of grade 2 astrocytoma or oligodendroglioma with a susceptible IDH1/2 mutation .
|
-

- HY-164476
-
|
|
EGFR
GSK-3
PD-1/PD-L1
|
Cancer
|
|
ES-072 is an orally effective selective EGFR mutant (EGFR-T790M) inhibitor. ES-072 activates GSK3α by inhibiting EGFR-T790M activity, which promotes phosphorylation of PD-L1 at Ser279 and Ser283. The phosphorylated PD-L1 recruits the E3 ubiquitin ligase ARIH1, leading to ubiquitination and proteasomal degradation of PD-L1. This mechanism not only reduces cancer cell growth but also enhances anti-tumor immune response by lowering PD-L1 levels. ES-072 can be used to inhibit proliferation in non-small cell lung cancer (NSCLC) cells .
|
-

- HY-174215
-
|
|
Tau Protein
|
Neurological Disease
|
|
TAU-IN-3 (Compound 2) is an orally active TAU inhibitor. TAU-IN-3 inhibits the expression of MAPT exon 10 DDPAC mutant gene in HeLa cells (IC50: 0.6 µM). TAU-IN-3 reduces the 4R/3R MAPT mRNA ratio in HeLa cells transfected with WT or DDPAC minigenes. TAU-IN-3 inhibits the insertion of endogenous MAPT exon 10 and the production of 4R tau protein in cells. TAU-IN-3 modulates tau splicing in htau mice and improves the associated behavioral phenotypes. TAU-IN-3 can be used to study neurodegenerative diseases .
|
-

- HY-161176
-
|
|
PROTACs
Ras
ERK
|
Cancer
|
|
PROTAC KRAS G12D degrader 1 is a selective PROTAC KRAS G12D degrader. PROTAC KRAS G12D degrader 1 inhibits proliferation of KRAS G12D-mutant cells and suppresses ERK phosphorylation. PROTAC KRAS G12D degrader 1 inhibits tumor growth in mice bearing AsPC-1 xenografts. PROTAC KRAS G12D degrader 1 can be used for the study of KRAS G12D-driven cancers.(Pink: KRAS ligand (HY-175892), Blue: VHL Ligand (HY-112078), Black: Linker, E3 ligase ligand-linker conjugate (HY-175893)) .
|
-

- HY-170968
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
EGFR-IN-150 is an EGFR inhibitor that effectively suppresses the phosphorylation of mutant epidermal growth factor receptor (EGFR) and its downstream AKT signaling pathway, thereby exerting antitumor effects and inducing HMOX1 expression to trigger ferroptosis. EGFR-IN-150 exhibits an IC50 of 0.386 μM against the non-small cell lung cancer (NSCLC) cell line H1975, and significantly inhibits colony formation and migration of both H1975 and A549 cells while inducing apoptosis. In addition, EGFR-IN-150 markedly suppresses tumor growth in the H1975 cell-derived xenograft (CDX) mouse model. EGFR-IN-150 holds promise for research related to non-small cell lung cancer .
|
-

- HY-E70739
-
|
|
c-Kit
|
Cancer
|
|
KIT (CD117) is an important cell surface marker used to identify certain types of hematopoietic(blood) progenitors in the bone marrow. KIT is a cytokine receptor expressed on the surface of hematopoietic stem cells as well as other cell types. Altered forms of this receptor may be associated with some types of cancer. KIT V559D/T670I is a mutant of KIT. KIT V559D/T670I Recombinant Human Active Protein Kinase is a recombinant KIT V559D/T670I protein that can be used to study KIT V559D/T670I-related functions .
|
-

- HY-403733C
-
|
|
Androgen Receptor
|
Cancer
|
|
JJ-450 is a non-competitive antagonist androgen receptor (AR) that inhibits the transcriptional activity of wild-type AR and mutant AR F876L. JJ-450 has an IC50 of approximately 1-10 μM in inhibiting AR transcriptional activity in PC3 cells. It is selective for AR binding and does not compete with androgens for binding to the ligand binding domain (LBD) of AR. JJ-450 inhibits the transcriptional activity of AR and its splice variants (such as AR F876L) by inhibiting AR nuclear translocation and promoting the degradation of unliganded AR in the nucleus. JJ-450 can be used in castration-resistant prostate cancer (CRPC) studies that are resistant to Enzalutamide (MDV3100) (HY-70003) .
|
-

- HY-169920
-
|
|
HIV
Reverse Transcriptase
|
Infection
|
|
HIV-1 inhibitor-79 (Compound 3k) is an HIV inhibitor that exhibits significant inhibitory activity against HIV-1 and its common mutant strains (with IC50 values of 1.9, 1.9, 8.7, and 11 nM against HIV-1, K103, L100I, and E138K, respectively), and has low cytotoxicity and a high selectivity index (CC50 = 21.95 μM, SI = 11478). Additionally, HIV-1 inhibitor-79 also shows antiviral activity against HIV-2, with an EC50 value of 6.14 μM, and significantly inhibits the activity of HIV-1 reverse transcriptase (IC50 = 25 nM) .
|
-

- HY-15800A
-
|
|
TNK1
LRRK2
|
Neurological Disease
Inflammation/Immunology
|
|
CZC-25146 is a potent and orally active LRRK2 inhibitor with IC50 values of 4.76 nM and 6.87 nM for wild-type LRRK2 and G2019S LRRK2, respectively. CZC-25146 inhibits PLK4, GAK, TNK1, CAMKK2 and PIP4K2C as well. CZC-25146 prevents mutant LRRK2-induced injury of neurons in vitro. CZC-25146 exhibits relatively favorable pharmacokinetic properties in mice. CZC-25146 can increase normal α-1-antitrypsin (AAT) secretion and reduce inflammatory cytokines. CZC-25146 can be used to research Parkinson's disease and liver diseases .
|
-

- HY-146223A
-
|
|
Ras
PI3K
p38 MAPK
Apoptosis
|
Cancer
|
|
(3R,10R,14aS)-AZD4625 is the isomer of AZD4625 (HY-146223), and can be used as an experimental control. AZD4625 is an orally active, selective irreversible, covalent allosteric GTPase KRASG12C inhibitor with an IC50 of 3 nM. AZD4625 can inhibit the MAPK pathway (with decreased pCRAF, pMEK, and pERK) and the PI3K pathway (with decreased pAKT and pS6), and induce cell apoptosis. AZD4625 has no binding and inhibition of wild-type RAS or isoforms carrying non-KRASG12C mutations. AZD4625 can be used for the study of KRASG12C mutant non-small cell lung cancer .
|
-

- HY-E70699
-
|
|
EGFR
|
Cancer
|
|
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR d746-750/T790M/C797S Recombinant Human Active Protein Kinase is a recombinant EGFR d746-750/T790M/C797S protein that can be used to study EGFR d746-750/T790M/C797S-related functions .
|
-

- HY-119396
-
|
|
Endogenous Metabolite
|
Cancer
|
|
DY3002 is a selective and highly potent EGFR inhibitor with activity in overcoming T790M-mediated drug resistance in non-small cell lung cancer. DY3002 exhibited superior inhibitory effects against EGFR T790M mutants in kinase assays (IC50 = 0.71 nM), compared to weaker inhibitory effects against wild-type EGFR (IC50 = 448.7 nM). DY3002 was significantly superior to rociletinib and osimertinib in selectivity, showing an extremely high selectivity index (SI = 632.0). In cell experiments, DY3002 had an IC50 value of 0.037 μM against H1975 cells, showing enhanced inhibitory potency. In addition, DY3002 was superior to other alternative compounds in terms of biological properties and did not cause hyperglycemia .
|
-

- HY-174453
-
|
|
PROTACs
Estrogen Receptor/ERR
Apoptosis
|
Cancer
|
|
PROTAC ERα Degrader-12 is a potent and selective Erα PROTAC degrader. PROTAC ERα Degrader-12 has antiproliferative effects in multiple breast cancer cell lines with wild-type or mutant ERα. PROTAC ERα Degrader-12 can halt the cell cycle and induce cell apoptosis. PROTAC ERα Degrader-12 exhibits excellent antitumor and ERα degradation activity. PROTAC ERα Degrader-12 can be used for research on breast cancer. (Pink: ER ligand-11 (HY-174475); Blue: VHL ligand (HY-112078); Black: Linker (HY-W088749); VHL ligand + Linker ( HY-W998310)) .
|
-

- HY-102069
-
|
|
Polo-like Kinase (PLK)
CDK
DAPK
|
Others
|
|
3MB-PP1, a bulky purine analog, is a Polo-like kinase 1 (Plk1) inhibitor. 3MB-PP1 blocks mitotic progression and cell division arise through target Plk1 in in cells expressing analog-sensitive Plk1 alleles. 3MB-PP1 specifically inhibits the activity of analog-sensitive Ssn3 (Cdk8). 3MB-PP1 inhibits Leu93 Mutant Zipper-interacting protein kinase (Leu93-ZIPK; IC50=2 μM). 3MB-PP1 can be used for the research of hypha formation of Candida albicans and cell division .
|
-

- HY-175297
-
|
|
VEGFR
EGFR
|
Cancer
|
|
EGFR T790M/VEGFR-2-IN-1 (Compound 6) is a dual EGFR T790M mutant (IC50=0.26 μM) and VEGFR-2 (IC50=0.95 μM) inhibitor. EGFR T790M/VEGFR-2-IN-1 blocks tumor cell proliferation and angiogenesis signaling pathways. EGFR T790M/VEGFR-2-IN-1 exhibits potent cytotoxicity against multiple cancer cell lines (HCT116, MCF-7, HepG2, A549; IC50=5.35-9.90 μM). EGFR T790M/VEGFR-2-IN-1 is promising for research of non-small cell lung cancer and solid tumors .
|
-

- HY-164552
-
|
|
Apoptosis
Androgen Receptor
|
Cancer
|
|
ZNU-IMB-Z15 (Compound Z15) is an antagonist of the androgen receptor (AR) and also a selective degrader of AR and ARV7. ZNU-IMB-Z15 can directly bind to the ligand-binding domain (LBD) and activation function-1 region of AR, and promote AR degradation through the proteasome pathway. ZNU-IMB-Z15 effectively inhibits the transcriptional activity of AR, AR mutants, and AR splice variants (ARVs), downregulating the mRNA and protein levels of AR downstream target genes, thereby overcoming the resistance to second-generation antiandrogen drugs induced by AR LBD mutations, AR amplification, and ARVs in castration-resistant prostate cancer (CRPC). ZNU-IMB-Z15 can inhibit the proliferation of AR-positive CRPC cell lines and induce their apoptosis, demonstrating anticancer activity both in vivo and in vitro .
|
-

- HY-174984
-
|
|
p97
Caspase
p62
|
Cancer
|
|
VCP/p97 IN-3 is a VCP/p97 allosteric inhibitor. VCP/p97 IN-3 shows the inhibitory activity against the VCP proteins with an IC50 of 9 nM and the mutant VCP proteins with IC50 of 12 nM (N660K) and 19 nM (V474A/D649A). VCP/p97 IN-3 increases K48-linked ubiquitination and the level of cleaved caspase-3. VCP/p97 IN-3 activates ER-stress and the UPR. VCP/p97 IN-3 inhibits tumor growth in RPMI-8226 cell subcutaneous xenograft mouse models. VCP/p97 IN-3 can be used for the study of multiple myeloma .
|
-

- HY-146019A
-
|
|
HIV
|
Infection
|
|
HIV-1 inhibitor-25 (compound R-12a) is a highly potent HIV-1 reverse transcriptase, with an IC50 value of 0.1061 μM. HIV-1 inhibitor-25 has high antiretroviral activity against WT HIV-1 with an EC50 of 13.6 nM, and exhibits relatively low cytotoxicity with a CC50 of 33.13 μM in MT-4 cells. HIV-1 inhibitor-25 also has inhibitory activity against HIV-1 mutant strains (L100I, K103N, Y181C, Y188L, E138K, F227L+V106A) with EC50 of 0.1961 ~ 5.8136 μM. HIV-1 inhibitor-25 can be used for researching AIDS .
|
-

- HY-159922
-
|
|
Androgen Receptor
|
Cancer
|
|
AR antagonist 9 is an orally bioavailable selective androgen receptor (AR) antagonist that exerts anticancer effects by disrupting the dimerization of AR ligand-binding domains, showing potential for overcoming drug resistance in prostate cancer (PCa). Its AR antagonistic activity has an IC50 value of 0.051 μM, comparable to Enzalutamide (HY-70002) (IC50 = 0.060 μM). AR antagonist 9 demonstrated superior efficacy against ARF876L/T877A and ARW741C mutants compared to Enzalutamide (HY-70002). Furthermore, AR antagonist 9 exhibited favorable pharmacokinetic properties, with an oral bioavailability of F = 66.24% in rats. In the LNCaP xenograft mouse model, oral administration of AR antagonist 9 significantly inhibited tumor growth. AR antagonist 9 holds promise for research into overcoming PCa drug resistance .
|
-

- HY-178938
-
|
|
Molecular Glues
Androgen Receptor
Caspase
Apoptosis
|
Endocrinology
Cancer
|
AR Degrader-3 is an orally active molecular glue that targets AR/ARV7 and induces the degradation of AR and ARV7 through the ubiquitin-proteasome pathway (UPP). AR Degrader-3 directly interacts with the ligand-binding domain (LBD) and the N-terminal domain (NTD) of AR. AR Degrader-3 effectively suppresses the transcriptional activity of wild-type AR (AR-WT), AR mutants, and ARV7. AR Degrader-3 downregulates the mRNA and protein levels of downstream AR target genes, thereby overcoming antiandrogen resistance mediated by ARV7 and AR point mutations. AR Degrader-3 induces apoptosis in Enzalutamide (HY-70002) (ENZa)-resistant cells and increases cleaved caspase-3 protein levels. AR Degrader-3 can be used for the study of castration-resistant prostate cancer (CRPC) .
|
-

- HY-169369
-
|
XJZ-06-462
|
MDM-2/p53
Apoptosis
|
Cancer
|
|
TRAP-1 (XJZ-06-462) is a selective p53 Y220C transcriptional activator. TRAP-1 can engage p53 Y220C and BRD4 in a ternary complex, which potently activates mutant p53 and triggers robust p53 target gene transcription. TRAP-1 rapidly upregulates p21 and other p53 target genes in pancreatic cell lines with p53 Y220C. TRAP-1 exhibits higher antiproliferative activity in BxPC-3 (p53 Y220C) cells than in A549 cells (p53 WT), with IC50 values of 0.531 μM and 3.94 μM, respectively. TRAP-1 can be used for the study of p53 Y220C-bearing cancer.
|
-

- HY-170926
-
|
|
RET
Apoptosis
|
Cancer
|
CQ1373 is a potent RET inhibitor, demonstrating cellular potency with IC50 values of 13.0, 25.7 and 28.4 nM against BaF3 cells expressing CCDC6-RET, CCDC6-RET-G810C and CCDC6-RET-G810R, respectively. CQ1373 exhibits good selectivity toward wild-type RET and solvent front mutants G810C/R with IC50 values of 4.2, 7.1 and 32.4 nM, respectively. CQ1373 inhibits RET phosphorylation and downstream signaling through SHC. CQ1373 induces Apoptosis and cell cycle arrest in BaF3 cells. CQ1373 exhibits anti-tumor efficacy and can be used for cancer research .
|
-

- HY-126077
-
MTI-31
1 Publications Verification
LXI-15029
|
mTOR
|
Inflammation/Immunology
Cancer
|
|
MTI-31 (LXI-15029) is a potent, orally active and highly selective inhibitor of mTORC1 and mTORC2. MTI-31 is selective for mTOR (Kd: 0.20 nM) versus PIK3CA, PIK3CB and PIK3G with >5,000 fold selectivity in mTOR binding assays. MTI-31 shows an IC50 of 39 nM for mTOR in LANCE assay of mTOR substrate phosphorylation with 100 μM ATP. MTI-31 can be used for the research of breast cancer .
|
-

- HY-D2946
-
|
BC-TMR
|
Fluorescent Dye
|
Infection
|
CLIP-TMR is a TMR-labeled CLIP tag fluorescent probe. CLIP-TMR combines the high specificity recognition ability of the CLIP-tag and the excellent optical performance of the TMR fluorophore, and can be used for the specific labeling and visualization of the HCV NS5A protein .
|
-

- HY-170911
-
|
|
PDGFR
|
Cancer
|
|
KIT/PDGFRA-IN-1 (compound 19) is a stem cell growth factor receptor (KIT) and platelet-derived growth factor receptor alpha (PDGFRA) inhibitor. KIT/PDGFRA-IN-1 inhibits KIT-wt, KIT-D816H, KIT-T670I, PDGFRA-wt, PDGFRA-D842V, PDGFRA-T674I and PDGFRA-G680R with IC50s of 2.3, 12, 492, 0.8, 99.9, 42.3, and 4.3 μM, respectively. KIT/PDGFRA-IN-1 inhibits GIST-T1, T1-a-D842V and GIST-48B cells (PDGFR- and KIT-Mutant GIST cell Lines) with GR50s of 12, 8900, ≥10 000 nM, respectively .
|
-

- HY-E70700
-
|
|
EGFR
|
Cancer
|
|
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR d746-750/T790M/C797S/L858R Recombinant Human Active Protein Kinase is a recombinant EGFR d746-750/T790M/C797S/L858R protein that can be used to study EGFR d746-750/T790M/C797S/L858R-related functions .
|
-

- HY-164490
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
LS-106 is an orally active and potent inhibitor against epidermal growth factor receptor (EGFR) . LS-106 exhibits antitumor activities both in vitro and in vivo. LS-106 inhibits the kinase activities of EGFR 19del/T790M/C797S and EGFR L858R/T790M/C797S with IC50 values of 2.4 nmol/L and 3.1 nmol/L, respectively, which is more potent than Osimertinib (HY-15772). LS-106 induces Apoptosis, suppresses cell proliferation of tumor cells harboring EGFR 19del/T790M/C797S and leas to significant tumor regression in a C797S-mutant xenograft model .
|
-

- HY-148369
-
|
|
PROTACs
Deubiquitinase
Apoptosis
MDM-2/p53
|
Cancer
|
|
U7D-1 is a first-in-class potent and selective USP7 (ubiquitin-specific protease 7) PROTAC degrader, with a DC50 of 33 nM in RS4;11 cells. U7D-1 shows anticancer activity. U7D-1 induces apoptosis in Jeko-1 cells .
|
-

- HY-178061
-
|
|
ERK
RET
|
Cancer
|
|
APS03118 is an orally active, potent and selective rearranged during transfection (RET) inhibitor. APS03118 broadly inhibits RET fusions and mutations (including G810, V804, L730, and Y806 variants), with IC50 values predominantly below 1 nM (0.095 nM for WT; ranging from 0.00438 to 5.72 nM for mutants), and demonstrates marked superiority against RET G810 mutations. APS03118 inhibits the entire RET signaling pathway (including RET, Shc, and ERK1/2), with >20-fold selectivity over most off-target kinases (except FLT3 and YES). APS03118 induces complete tumor regression in KIF5B-RET and CCDC6-RET V804 M patient derived xenografts (PDXs) and significantly prolongs survival in an intracranial CCDC6-RET metastasis mice model. APS03118 can be used for selective RET inhibitor (SRI)-resistant, RET-driven cancer research .
|
-

- HY-164392
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
TAS-121 is an orally active, selective, covalent, third-generation mutant EGFR-tyrosine kinase inhibitor (EGFR-TKI). TAS-121 inhibits the L858R mutation (IC50=1.7 nM), Ex19del mutation (IC50=2.7 nM), L858R/T790M mutation (IC50=0.56 nM) and Ex19del/T790M mutation (IC50=1.1 nM) and wild-type EGFR (IC50=8.2 nM). TAS-121 inhibits HER2 and HER4 with IC50s of 110 and 2.6 nM, respectively. TAS-121 inhibits phosphorylation of EGFR and its downstream signaling targets to block cell proliferation. TAS-121 induces apoptosis and displays antitumor activity in SW48 (EGFR G719S) and NCI-H1975 (EGFR L858R/T790M) xenograft models .
|
-

- HY-153803
-
|
|
PROTACs
Molecular Glues
Btk
|
Cancer
|
GBD-9 is a degrader based on the E3 ubiquitin ligase CRBN that targets BTK and the G1 to S phase transition protein GSPT1. GBD-9 has both PROTAC and molecular glue properties by inducing ubiquitination and proteasomal degradation of target proteins. GBD-9 can efficiently degrade wild-type and mutant BTK (such as C481S mutation) and GSPT1. GBD-9 significantly inhibits tumor cell proliferation by inducing G1 phase arrest in cancer cells, downregulating anti-apoptotic proteins (BCL-2, MCL-1) and activating Caspase-3 to induce apoptosis. GBD-9 is mainly used in the research of hematological tumors such as diffuse large B-cell lymphoma (DLBCL) and acute myeloid leukemia (AML) .
GBD-9 is composed of E3 ubiquitin ligase ligand (pink part) 5-Aminothalidomide (HY-W023573), target protein ligand (blue part) Btk Inhibitor: IBT6A (HY-13036A), and PROTAC linker (black part) Nonanoic acid (HY-N7057).
|
-

- HY-169385
-
|
|
AUTOTACs
Androgen Receptor
|
Cancer
|
|
ATC-324 is an bivalent AR (Androgen Receptor) degrader based on the protein degradation technology platform AUTOphagy-TArgeting Chimera (AUTOTAC). ATC-324 induces the formation of AR/p62 complex, leading to autophagy-lysosomal degradation of AR. ATC-324 can reduce nuclear AR levels and downregulate target gene expression of AR and AR-v7, and also has a degradation effect on common AR mutants in PCa . ATC-324 is composed of target-binding ligand (TBL) Enzalutamide (HY-70002) and p62/SQSTM1 autophagy-targeting ligand (ATL) YT 6-2 analog-1 (HY-169386), connected by Boc-NH-PEG4-CH2CH2NH2 (HY-W008352). Among them, the active control of the target protein ligand is Enzalutamide carboxylic acid (HY-70002B), and the conjugate composed of the autophagy-targeting ligand and the linker is YT 6-2-PEG3-C2-NH2 (HY-169387).
|
-

- HY-163679
-
|
|
Estrogen Receptor/ERR
Cytochrome P450
PROTACs
Apoptosis
|
Cancer
|
|
PROTAC ERα Degrader-9 (Compound 18c) is a dual-targeting PROTAC degrader, which degrades estrogen receptor α (ERα) and aromatase (ARO). PROTAC ERα Degrader-9 binds to ERα with a Ki of 0.25 μM, inhibits ARO with an IC50 of 4.6 μM. PROTAC ERα Degrader-9 inhibits the proliferation of MCF-7 wildtype (IC50=0.54 μM) and ERα mutants MCF-7 EGFR (IC50=0.075 μM), MCF-7 D538G (IC50=0.31 μM), MCF-7 Y537S (IC50=2.3 μM), downregulates the expressions of ERS1 and MYC. PROTAC ERα Degrader-9 arrests the cell cycle at G2/M, induces apoptosis in MCF-7. PROTAC ERα Degrader-9 exhibits antitumor efficacy in mouse models. (Pink: ligand for target protein (HY-163680); Black: linker (HY-W007559); Blue: ligand for E3 ligase (HY-112078))
|
-

- HY-175610
-
|
|
PROTACs
FLT3
JAK
Epigenetic Reader Domain
|
Cancer
|
|
PROTAC FLT3/JAK2/BRD4 Degrader-1 is a PROTAC degrader that target FLT3, JAK2, and BRD4 with DC50 values of 5.23, 0.678, and 1.17 nM, respectively. PROTAC FLT3/JAK2/BRD4 Degrader-1 exhibits potent antiproliferative activity against MV4;11 cells (IC50 = 0.79 nM) and FLT3 mutant-transformed Ba/F3 cells. PROTAC FLT3/JAK2/BRD4 Degrader-1 induces apoptosis in MV4;11 cells. PROTAC FLT3/JAK2/BRD4 Degrader-1 demonstrates significant anti-tumor efficacy in the MV4;11 xenograft model established in NOD SCID mice. PROTAC FLT3/JAK2/BRD4 Degrader-1 can be used for the study of acute myeloid leukemia (AML). (Pink: FLT3/JAK2/BRD4 ligand (HY-175611), Blue: CRBN Ligand (HY-W087383), Black: Linker, E3 ligase ligand-linker conjugate (HY-W897939)) .
|
-

| Cat. No. |
Product Name |
Type |
-
- HY-114491
-
|
|
Fluorescent Dye
|
|
Rineterkib (compound B) is an orally available ERK1 and ERK2 inhibitor in the treatment of a proliferative disease characterized by activating mutations in the MAPK pathway. The activity is particularly related to the treatment of KRAS-mutant NSCLC, BRAF-mutant NSCLC, KRAS-mutant pancreatic cancer, KRAS-mutant colorectal cancer (CRC) and KRAS-mutant ovarian cancer. Rineterkib hydrochloride can also inhibit RAF .
|
-
- HY-W127705
-
|
|
Fluorescent Dye
|
|
Quinacrine mustard dihydrochloride is a fluorochrome. Quinacrine mustard dihydrochloride as a polycyclic aromatic agent can be used as mutagenic agent induces the mutants of bacteria. Quinacrine mustard dihydrochloride induces cell cycle arrest at G2/M-phase. Quinacrine mustard dihydrochloride has the potential for the research of plant, animal, or human chromosomes .
|
-
- HY-156712
-
|
|
Fluorescent Dye
|
|
Depatuxizumab MMAE is an antibody-drug conjugate (ADC) comprising an anti EGFR monoclonal antibody (Depatuxizumab) (HY-P99849) and the cytotoxic agent Monomethyl auristatin E (MMAE) (HY-15162). Depatuxizumab MMAE can be used for the study of EGFR-expressing advanced solid tumors.
|
-
- HY-143218
-
TPE-MI
Maximum Cited Publications
9 Publications Verification
Tetraphenylethene maleimide
|
Fluorescent Dye
|
|
TPE-MI (Tetraphenylethene maleimide) is a thiol probe for measuring unfolded protein load and proteostasis in cells (the excitation wavelength is 350 nm and the emission wavelength is 470 nm). TPE-MI can report imbalances in proteostasis in induced pluripotent stem cell models of Huntington disease, as well as cells transfected with mutant Huntington exon 1 before the formation of visible aggregates. TPE-MI also detects protein damage following dihydroartemisinin research of the malaria parasitesPlasmodium falciparum .
|
-
- HY-D0098
-
|
N-(5-Fluoresceinyl)maleimide
|
Fluorescent Dye
|
Fluorescein-5-maleimide (N-(5-Fluoresceinyl)maleimide) is a fluorescent dye. Fluorescein-5-maleimide can be used to detect the redox state of thiols in eukaryotic cells. Fluorescein-5-maleimide can label peptides and is used to detect negatively charged nanoparticles. Fluorescein-5-maleimide can also label actin to explore its interaction with cardiac myosin-binding protein C (cMyBP-C), which helps in developing small molecule modulators for heart failure. Fluorescein-5-maleimide can screen mutant proteins that contain cysteine residues. The excitation wavelength of Fluorescein-5-maleimide is 494 nm, and the emission wavelength is 519 nm .
|
-
- HY-19980G
-
|
APR-246 (GMP); PRIMA-1Met (GMP)
|
Fluorescent Dye
|
|
Eprenetapopt (GMP) (APR-246(GMP)) is Eprenetapopt (HY-19980) in GMP grade. Eprenetapopt (APR-246) is a first-in-class, small molecule that restores wild-type p53 functions in TP53-mutant cells. Eprenetapopt triggers apoptosis in tumor cells. Eprenetapopt also targets the selenoprotein thioredoxin reductase 1 (TrxR1), a key regulator of cellular redox balance .
|
-
- HY-DY1024
-
|
|
Fluorescent Dye
|
TPE-MI (Tetraphenylethene maleimide) (solution) is a thiol probe for measuring unfolded protein load and proteostasis in cells (the excitation wavelength is 350 nm and the emission wavelength is 470 nm). TPE-MI can report imbalances in proteostasis in induced pluripotent stem cell models of Huntington disease, as well as cells transfected with mutant Huntington exon 1 before the formation of visible aggregates. TPE-MI also detects protein damage following dihydroartemisinin research of the malaria parasitesPlasmodium falciparum .
Solution Concentration: 10 mM
|
-
- HY-D2946
-
|
BC-TMR
|
Fluorescent Dye
|
CLIP-TMR is a TMR-labeled CLIP tag fluorescent probe. CLIP-TMR combines the high specificity recognition ability of the CLIP-tag and the excellent optical performance of the TMR fluorophore, and can be used for the specific labeling and visualization of the HCV NS5A protein .
|
| Cat. No. |
Product Name |
Type |
-
- HY-D1056C5
-
|
LPS, from Salmonella enterica (Serotype minnesota Re 595 (Re mutant))
|
Biochemical Assay Reagents
|
Lipopolysaccharides (LPS), from S. enterica (Salmonella enterica) serotype minnesota Re 595 (Re mutant) is prepared from Salmonella enterica strain Re 595 (Re mutant). The structure in the LPS of strain Re 595 was shown to induce secretion and aggregation in human platelets .
It is recommended to prepare a solution with concentration ≥2 mg/mL. Vortex thoroughly for more than 10 minutes. Due to the adsorption characteristics of LPS, silanized container or low adsorption centrifuge tubes should be used for aliquoting and storage, and mix thoroughly before use.
|
-
- HY-W004705
-
|
2-Hydroxymethylaniline
|
Biochemical Assay Reagents
|
|
(2-Aminophenyl)methanol (2-Hydroxymethylaniline) is a molecular chaperone to rescue P123S mutant pendrin. (2-Aminophenyl)methanol has the advantages of low dose, long-term effect and low toxicity. (2-Aminophenyl)methanol can be used for the study of Pendred syndrome (a syndromic deafness) .
|
-
- HY-W007626
-
|
|
Biochemical Assay Reagents
|
|
3,5-Dimethoxybenzaldehyde is an antifungal agent. 3,5-Dimethoxybenzaldehyde exhibits antifungal activity against Saccharomyces cerevisiae cell wall integrity mutants (slt2Δ and bck1Δ) and Aspergillus fumigatus MAPK mutants (sakAΔ and mpkCΔ) .
|
-
- HY-W002798
-
|
|
Biochemical Assay Reagents
|
|
Cyclopropanesulfonyl chloride is an intermediate. Cyclopropanesulfonyl chloride can be used to synthesize Compound 1. Compound 1 has anticancer activity against EGFR triple-mutant tumors .
|
-
- HY-145411
-
|
|
Biochemical Assay Reagents
|
|
PEG2000-C-DMG, a pegylated lipid, can be used for the preparation of Onpattro. Onpattro, a hepatically directed investigational RNAi therapeutic agent, harnesses this process to reduce the production of mutant and wild-type transthyretin by targeting the 3′ untranslated region of transthyretin mRNA .
|
-
- HY-59219
-
|
|
Biochemical Assay Reagents
|
|
Cyclopropylboronic acid is an intermediate. Cyclopropylboronic acid can be used to synthesize Compound 25. Compound 25 has antiproliferative effects on EGFR mutant (EGFR Δ19del/T790M/C797S) cells. Cyclopropylboronic acid can be used in lung cancer research .
|
-
- HY-114365
-
|
UDP-N-acetyl-D-galactosamine disodium
|
Biochemical Assay Reagents
|
|
UDP-GalNAc (UDP-N-acetyl-D-galactosamine) disodium is a sugar nucleotide and a substrate for EpsC115. EpsC115 is a mutant with N-terminal residues 1-115 deleted from the exopolymeric substance (EPS). UDP-GalNAc disodium is a donor substrate for many N-acetylgalactosaminyltransferases, which transfer GalNAc from nucleotide sugars to sugar or peptide acceptors. UDP-GalNAc disodium provides a sugar group donor for glycosylation reactions. UDP-GalNAc disodium can be used in cancer research, such as colorectal and breast cancer .
|
-
- HY-19980G
-
|
APR-246 (GMP); PRIMA-1Met (GMP)
|
Biochemical Assay Reagents
|
|
Eprenetapopt (GMP) (APR-246(GMP)) is Eprenetapopt (HY-19980) in GMP grade. Eprenetapopt (APR-246) is a first-in-class, small molecule that restores wild-type p53 functions in TP53-mutant cells. Eprenetapopt triggers apoptosis in tumor cells. Eprenetapopt also targets the selenoprotein thioredoxin reductase 1 (TrxR1), a key regulator of cellular redox balance .
|
-
- HY-116762
-
|
|
Biochemical Assay Reagents
|
|
Quorum sensing is a regulatory system used by bacteria to control gene expression in response to increased cell density. The control of bacterial infection by quenching the quorum sensing system of bacteria is a promising research area. The expression of specific target genes, such as transcriptional regulators belonging to the LuxIR protein family, is coordinated by the synthesis of diffusible acyl homoserine lactone (AHL) molecules. N-butyryl-L-Homocysteine thio-lactone is an analog of N-butyryl-L-homoserine lactone, a small, diffusible signaling molecule involved in quorum sensing, thereby controlling gene expression and cellular metabolism . N-butyryl-L-homocysteine thiolactone induces violacein expression in Viola viola mutants that normally fail to produce AHL.
|
-
- HY-W745090
-
|
|
Biochemical Assay Reagents
|
|
Isomaltulose monohydrate is a fMLP inhibitor and also inhibits Src kinase, ERK1/2, p38 and AKT phosphorylation signals in immune regulation. Isomaltulose monohydrate can interfere with the interaction between the βγ subunit of the fMLP receptor Gi protein and its downstream molecules, thereby inhibiting fMLP-induced respiratory burst. Isomaltulose monohydrate inhibits fMLP (0.1 μM)-induced superoxide anion production (IC50: 1.98 μM) , cathepsin G release (IC< sub>50: 2.76 μM) and chemotaxis. Isomaltulose monohydrate can improve excessive activation of neutrophils and reduce inflammation or tissue damage. A series of derivatives of Isomaltulose monohydrate are found to have inhibitory effects on FSGS-related TRPC6 functional mutants .
|
| Cat. No. |
Product Name |
Target |
Research Area |
-
- HY-P2361
-
|
|
Ras
|
Others
|
|
S12 is a mutant RAS peptide containing the Gly (G) to Ser (S12) substitution. The sequence of the peptide is KLVVVGASGVGKS .
|
-
- HY-P2360
-
|
Ras 5-17
|
Ras
|
Others
|
|
G12 (Ras 5-17) is a wild-type Ras peptide consisted of amino acids 5-17 (KLVVVGAGGVGKS). G12 can be used as a control of mutant Ras peptides studies (such V12) .
|
-
- HY-P5082
-
|
|
α-synuclein
|
Neurological Disease
|
|
α-Synuclein 4554W is an inhibitor of α-Synuclein (aSyn) aggregation with associated toxicity. α-Synuclein 4554W consists of GIVNGVKA sequences, previously identified through intracellular library screening. α-Synuclein 4554W reduces fibril formation of aSyn mutants assocaited with Parkinson’s disease .
|
-
- HY-P2265A
-
|
|
SOS1
Ras
|
Cancer
|
|
SAH-SOS1A TFA is a peptide-based SOS1/KRAS protein interaction inhibitor. SAH-SOS1A TFA binds to wild-type and mutant KRAS (G12D, G12V, G12C, G12S, and Q61H) with nanomolar affinity (EC50=106-175 nM). SAH-SOS1A TFA directly and independently blocks nucleotide association. SAH-SOS1A TFA impairs KRAS-driven cancer cell viability and exerts its effects by on-mechanism blockade of the ERK-MAPK phosphosignaling cascade downstream of KRAS .
|
-
- HY-P2670
-
|
|
NF-κB
|
Others
|
|
SN50M, a mutant peptide of SN50 (HY-P0151), is a cell membrane-permeable inactive control peptide .
|
-
- HY-P2250
-
|
ELA-32 negative control
|
Apelin Receptor (APJ)
|
Others
|
|
ELA RR>GG (ELA-32 negative control), an ELABELA (ELA-32 human) mutant peptide, is inactive. ELA RR>GG is a negative control for ELABELA (HY-P2196) .
|
-
- HY-P2360A
-
|
Ras 5-17 TFA
|
Ras
|
Others
|
|
G12 (Ras 5-17) TFA is a wild-type Ras peptide consisted of amino acids 5-17 (KLVVVGAGGVGKS). G12 TFA can be used as a control of mutant Ras peptides studies (such V12) .
|
-
- HY-P10425
-
|
|
Histone Methyltransferase
|
Cancer
|
|
T2857W is a mutant peptide that has inhibitory effect on KIX-MLL interaction (IC50=5.67 μM). T2857W can be used for protein-protein interaction (PPI) and for the study of diseases related to KIX-MLL interaction, such as leukemia, cancer, etc .
|
-
- HY-P2265
-
|
|
SOS1
Ras
|
Cancer
|
|
SAH-SOS1A is a peptide-based SOS1/KRAS protein interaction inhibitor. SAH-SOS1A binds to wild-type and mutant KRAS (G12D, G12V, G12C, G12S, and Q61H) with nanomolar affinity (EC50=106-175 nM), directly and independently blocks nucleotide association, impairs KRAS-driven cancer cell viability, and exerts its effects by on-mechanism blockade of the ERK-MAPK phosphosignaling cascade downstream of KRAS .
|
-
- HY-P10456
-
|
|
Peptides
|
Inflammation/Immunology
|
|
[Leu144,Arg147]-PLP (139-151) is a mutant peptide fragment of myelin proteolipid protein (PLP), with the tryptophan and histidine at positions 144 and 147 respectively replaced by leucine and arginine. [Leu144,Arg147]-PLP (139-151) also serves as a T cell receptor (TCR) antagonist for encephalitogenic Th1 clones, blocking their activation in vitro. Furthermore, [Leu144,Arg147]-PLP (139-151) can inhibit the development of experimental autoimmune encephalomyelitis (EAE) .
|
-
- HY-129028
-
-
- HY-129028A
-
|
|
Peptides
|
Others
|
|
H-Ala-Ala-Tyr-OH TFA can be synthesized mutant peptides .
|
-
- HY-P5192
-
|
|
Ras
|
Cancer
|
|
KRAS G12D 8-16 is a mutant KRAS G12D 8-16 peptide .
|
-
- HY-P11089
-
|
|
MHC
|
Cancer
|
|
TP53 neoepitope is a high-affinity antigenic peptide targeting HLA-A. TP53 neoepitope can triggers CD8 + T cell-mediated killing of TP53-mutant tumor cells. TP53 neoepitope is promising for research of solid tumors harboring TP53 hotspot mutations (e.g., R175H, R273H) .
|
-
- HY-P3486
-
-
- HY-P3488
-
-
- HY-P0222
-
|
|
PKA
|
Others
|
|
PKI(5-24) is a potent, competitive, and synthetic peptide inhibitor of PKA (cAMP-dependent protein kinase), with a Ki of 2.3 nM. PKI(5-24) corresponds to residues 5-24 in the naturally occurring heat-stable protein kinase inhibitor .
|
-
- HY-P0222A
-
|
|
PKA
|
Others
|
|
PKI(5-24) TFA is a potent, competitive, and synthetic peptide inhibitor of PKA (cAMP-dependent protein kinase), with a Ki of 2.3 nM. PKI(5-24) TFA corresponds to residues 5-24 in the naturally occurring heat-stable protein kinase inhibitor .
|
-
- HY-P3277
-
|
|
Ras
|
Cancer
|
|
KRpep-2d is a potent K-Ras inhibitor and inhibits proliferation of K-Ras-driven cancer cells. KRpep-2d can be used for cancer research .
|
-
- HY-P3277A
-
|
|
Ras
|
Cancer
|
|
KRpep-2d (TFA) is a potent K-Ras inhibitor and inhibits proliferation of K-Ras-driven cancer cells. KRpep-2d can be used for cancer research .
|
-
- HY-P4391
-
-
- HY-P2319
-
|
|
p38 MAPK
JNK
|
Inflammation/Immunology
|
|
OVA-E1 peptide, is an antagonist variant of SIINFEKL [OVA (257-264). OVA-E1 peptide, activates the p38 and JNK cascades similarly in mutant and wild-type thymocytes .
|
-
- HY-P2319A
-
|
|
p38 MAPK
JNK
|
Inflammation/Immunology
|
|
OVA-E1 peptide TFA, is an antagonist variant of SIINFEKL [OVA (257-264). OVA-E1 peptide, activates the p38 and JNK cascades similarly in mutant and wild-type thymocytes .
|
-
- HY-P1974
-
|
WF11899A
|
Antibiotic
|
Infection
|
|
FR901379 (WF11899A) is a novel echinocandin-type lipopeptide antibiotic. FR901379 is primarily produced by mutant strain M-7 of Coleophoma empetri F-11899 (FERM BP-2635) .
|
-
- HY-P5968
-
|
β(25-35)KA
|
Amyloid-β
|
Neurological Disease
|
|
[Ala28]-β Amyloid(25-35) (β(25-35)KA) is an electrically neutral mutant peptide of Aβ(25-35) that accelerates the aggregation of Firefly Luciferase .
|
-
- HY-P10610
-
|
|
MDM-2/p53
|
Cancer
|
|
Peptide 234CM is a peptide containing isoleucine at position 3, corresponding to the sequence of a point mutation in p53 codon 234. Peptide 234CM induces potent cytotoxic T cell (CTL) and antitumor immune responses against mutant p53 .
|
-
- HY-P5968A
-
|
β(25-35)KA TFA
|
Amyloid-β
|
Neurological Disease
|
|
[Ala28]-β Amyloid(25-35) (β(25-35)KA) (TFA) is an electrically neutral mutant peptide of Aβ(25-35) that accelerates the aggregation of Firefly Luciferase .
|
-
- HY-P10610A
-
|
|
MDM-2/p53
|
Cancer
|
|
Peptide 234CM TFA is a peptide containing isoleucine at position 3, corresponding to the sequence of a point mutation in p53 codon 234. Peptide 234CM TFA induces potent cytotoxic T cell (CTL) and antitumor immune responses against mutant p53 .
|
-
- HY-P4528
-
|
|
Ser/Thr Protease
|
Others
|
|
Ala-Phe-Pro-βNA is the subsrate of prolyl tripeptidyl aminopeptidase, which is from Porphyromonas gingivalis. Ala-Phe-Pro-βNA binds to H-Ala-Ile-pyrrolidin-2-yl boronic acid for PTP39 and the E636A mutant with Ki values of 88.1 nM and 48.8 nM, respectivley .
|
-
- HY-P10438
-
|
|
Raf
|
Cancer
|
|
TAT-Braftide is a peptide inhibitor designed to block the dimerization of BRAF, thereby inhibiting its kinase activity. The destruction of BRAF dimer by TAT-Braftide makes BRAF protein more susceptible to proteasome degradation, directly inhibits the activity of BRAF kinase, and reduces the activation of MAPK signaling pathway. Tat-braftide can be used for the role of RAF kinase in MAPK signaling pathway and for the study of BRAF mutant cancers .
|
-
- HY-P11356
-
|
|
IFNAR
|
Cancer
|
|
KRAS G12V Peptide is a specific peptide derived from the Kirsten rat sarcoma virus (KRAS) gene carrying the G12V oncogenic mutation. KRAS G12V Peptide induces responses like IFN-γ secretion and cytotoxicity. KRAS G12V Peptide can be used for the study of immune responses against KRAS G12V-mutant tumors .
|
-
- HY-P11357
-
|
|
IFNAR
|
Cancer
|
|
KRAS G12C Peptide is a specific peptide derived from the Kirsten rat sarcoma virus (KRAS) gene carrying the G12C oncogenic mutation. KRAS G12C Peptide induces responses like IFN-γ secretion and cytotoxicity. KRAS G12C Peptide can be used for the study of immune responses against KRAS G12C-mutant tumors .
|
-
- HY-P10436
-
|
|
Raf
|
Cancer
|
|
Braftide is an allosteric inhibitor for BRAF kinase by targeting the dimer interface of BRAF kinase and inhibiting the formation of BRAF dimers. Braftide inhibits wild-type BRAF and oncogenic BRAF G469A with IC50 of 364 nM and 172 nM, respectively. Braftide inhibits MAPK signaling pathway, inhibits proliferation of KRAS mutant tumor cells (EC50 is 7.1 and 6.6 μM, for HCT116 and HCT-15), in combination of TAT sequence. Braftide can be used for cancer research .
|
-
- HY-P5395
-
|
|
HIV
|
Others
|
|
TAT-GluR23A Fusion Peptide is a biological active peptide. (This is the GluR23A sequence, a control inactive peptide used as a mutant counterpart to glutamate receptor endocytosis inhibitor (GluR23Y), connected to an 11 amino acid cell permeable HIV Trans-Activator of Transcription (TAT) protein transduction domain (PTD). GluR23A is derived from GluR23Y amino acids 869 to 877, with Ala substituted for Tyr, and thus lacking essential phosphorylation sites.Control peptide of HY-P2259)
|
-
- HY-P11357A
-
|
|
IFNAR
|
Cancer
|
|
KRAS G12C Peptide TFA is the trifluoroacetate salt of KRAS G12C Peptide (HY-P11357). KRAS G12C Peptide is a specific peptide derived from the Kirsten rat sarcoma virus (KRAS) gene carrying the G12C oncogenic mutation. KRAS G12C Peptide induces responses like IFN-γ secretion and cytotoxicity. KRAS G12C Peptide can be used for the study of immune responses against KRAS G12C-mutant tumors .
|
-
- HY-P11356A
-
|
|
IFNAR
|
Cancer
|
|
KRAS G12V Peptide TFA is the trifluoroacetate salt of KRAS G12V Peptide (HY-P11355). KRAS G12V Peptide is a specific peptide derived from the Kirsten rat sarcoma virus (KRAS) gene carrying the G12V oncogenic mutation. KRAS G12V Peptide induces responses like IFN-γ secretion and cytotoxicity. KRAS G12V Peptide can be used for the study of immune responses against KRAS G12V-mutant tumors .
|
-
- HY-P10976
-
|
|
SARS-CoV
|
Infection
|
|
Spike Glycoprotein (1147-1162) is a linear and broadly neutralizing peptide in the S2 protein of SARS-CoV-2. Spike Glycoprotein (1147-1162) is conserved across SARS-CoV, BatCoV RaTG13, SARS-CoV-2, and SARS-CoV-2 variants. Spike Glycoprotein (1147-1162)-targeting mAbs can neutralize both SARS-CoV-2 and SARS-CoV by preventing fusion between the virus and cell membrane. Spike Glycoprotein (1147-1162) can be used for universal vaccines against SARS-CoV-2 mutants research .
|
-
- HY-K0233
-
|
|
|
MCE Anti-RFP Affinity Gel can be used for the detection and purification of native RFP, RFP mutants, and IP assays.
|
-
- HY-KE8004
-
1 Publications Verification
|
|
Reverse Transcriptase is a reverse transcriptase that clones and expresses the deletion mutant RNase H- of M-MuLV through genetic recombination technology.
|
| Cat. No. |
Product Name |
Target |
Research Area |
-
- HY-P99849
-
|
ABT-806
|
EGFR
|
Cancer
|
|
Depatuxizumab is a brain-penetrant and humanized tumor-specific anti EGFR monoclonal antibody. Depatuxizumab inhibits the growth of xenograft models of mutant EGFRvIII and wild-type EGFR. Depatuxizumab can be used for research on cancer .
|
-
- HY-P99701
-
|
BMS-986004
|
TNF Receptor
|
Inflammation/Immunology
|
|
Letolizumab (BMS-986004) is a monoclonal antibody targeting CD40L, which is produced to express mutant IgG1 lacking effector function, including Fc binding and complement fixation. Letolizumab reduces rejection, thromboembolism and prolongs the survival time .
|
-
- HY-P99823
-
|
TA-46; sFGFR3
|
FGFR
|
Others
|
|
Recifercept (TA-46) is a soluble, recombinant fibroblast growth factor receptor 3 (FGFR3) molecule. Recifercept can be used as a decoy/ligand trap to decrease the amount of fibroblast growth factors that can bind to mutant FGFR3 receptors. Recifercept can be used for the research of achondroplasia .
|
-
- HY-P99934
-
|
ABBV-621
|
Apoptosis
Caspase
TNF Receptor
|
Cancer
|
|
Eftozanermin alfa (ABBV-621) is a tumor necrosis factor-related apoptosis-inducing ligand receptor (TRAIL-R) agonist. Eftozanermin alfa is a fusion protein consisting of a mutant immunoglobulin G1-Fc linked to 2 single-chain trimers of TRAIL. Eftozanermin alfa induces apoptosis in tumor cells by activation of death receptors (DR4 receptor and DR5 receptor) with Kds of 780 nM and 635 nM. Eftozanermin alfa can be used for the research of multiple solid and heme malignancies .
|
-
- HY-P991571
-
|
GC-1118A
|
EGFR
PERK
Akt
|
Cancer
|
|
GC1118 (GC-1118A) is a fully human anti-EGFR monoclonal antibody with binding affinity of 0.16 nM (KD) to EGFR. GC1118 displays potent inhibitory effects on high- and low-affinity EGFR ligand-induced signaling. GC1118 shows potent anti proliferative activity in KRAS wild-type and KRAS mutant cells. GC1118 can reach the tumor by crossing both BBB (blood-brain barrier) and BTB (brain-tumor barrier) and shows superior anti-tumor effects in various mice xenograft models. GC1118 can be used for the researches of cancer, such as colorectal cancer .
|
| Cat. No. |
Product Name |
Category |
Target |
Chemical Structure |
-
- HY-N3492
-
-
-
- HY-N3920
-
-
-
- HY-N5141
-
-
-
- HY-121262
-
|
Bisabolene; γ-Bisabolene
|
Other Terpenoids
Microorganisms
Terpenoids
Source classification
|
Others
|
|
Bisabolene is a compound that can be synthesized by engineered bacteria. Bisabolene can be synthesized by heterologous expression of related genes in cyanobacteria. Its production and changes in related metabolites were studied in glycogen-deficient mutants.
|
-
-
- HY-W746957
-
-
-
- HY-W753726
-
-
-
- HY-N3496
-
-
-
- HY-N16232
-
|
2-α-Carboxy cholestanol
|
Lipid
|
Hedgehog
|
|
2-ACC (2-α-Carboxy cholestanol) is a cholestanol, is a mutant Sonic Hedgehog protein D46A activator with an EC50 of ~5 μM.
|
-
-
- HY-120885
-
-
-
- HY-N14782
-
-
-
- HY-P1974
-
-
-
- HY-B0282
-
-
-
- HY-N0328
-
-
-
- HY-114604
-
|
|
Natural Products
Microorganisms
Source classification
|
Bacterial
|
|
Propioxatin B is a tricyclic sesquiterpenoid compound isolated from the root of vetiver grass. It has anti-tuberculosis activity and inhibitory effects on a variety of drug-resistant mutants of Mycobacterium tuberculosis. In computer simulation docking studies, it showed binding affinity with bacterial DNA gyrase and has a certain safety in vivo.
|
-
-
- HY-122958
-
-
-
- HY-B0282R
-
|
ACh chloride (Standard)
|
Natural Products
Source classification
Endogenous metabolite
|
Reference Standards
nAChR
Calcium Channel
Endogenous Metabolite
|
|
Acetylcholine (chloride) (Standard) is the analytical standard of Acetylcholine (chloride). This product is intended for research and analytical applications. Acetylcholine chloride (ACh chloride), a neurotransmitter, is a potent cholinergic agonist. Acetylcholine chloride is a modulator of the activity of dopaminergic (DAergic) neurons through the stimulation of nicotinic acetylcholine receptors (nAChRs) . Acetylcholine chloride inhibits p53 mutant peptide aggregation in vitro .
|
-
-
- HY-I0960
-
-
-
- HY-N7484
-
-
-
- HY-N12390
-
-
-
- HY-125540
-
-
-
- HY-137971
-
|
|
Microorganisms
Ketones, Aldehydes, Acids
Source classification
|
Ras
|
|
(±)-Spiro-oxanthromicin A (Compound 4), a polyketide, is a K-Ras inhibitor with an IC50 of 26.7 μM. (±)-Spiro-oxanthromicin A can be isolated from soil-derived Streptomyces sp. (±)-Spiro-oxanthromicin A can mislocalize oncogenic mutant K-Ras from the plasma membrane of intact MDCK cells. (±)-Spiro-oxanthromicin A can be used for cancers research .
|
-
-
- HY-N6790
-
-
-
- HY-N0328R
-
-
-
- HY-130259
-
-
-
- HY-N12109
-
|
|
Microorganisms
Ketones, Aldehydes, Acids
Source classification
|
Others
|
|
Albizziin is an amino acid analog with activity as a competitive inhibitor of asparagine synthetase. Albizziin has been used to isolate Chinese hamster ovary cell mutants with altered levels of the target enzyme. Several mutational classes of albizziin can be distinguished based on cross-resistance to β-aspartic hydroxamic acid. Studies on asparagine synthetase have shown that resistance to albizziin may be associated with altered regulation of asparagine synthetase, structural mutations of the enzyme, and gene amplification .
|
-
-
- HY-122936
-
-
-
- HY-114365
-
-
-
- HY-N2099
-
-
-
- HY-N2099A
-
|
(Z)-Senegin III
|
Structural Classification
Polygala senega L.
Polysaccharides
Source classification
Polygalaceae
Plants
Saccharides
|
Drug Derivative
|
|
(Z)-Onjisaponin B ((Z)-Senegin III) is a derivative of Onjisaponin B (HY-N2099). Onjisaponin B is an orally active natural product derived from Polygala tenuifolia. Onjisaponin B inhibits NF-κB p65. Onjisaponin B enhances autophagy and accelerates the degradation of mutant α-synuclein and huntingtin. Onjisaponin B reduces β-amyloid (Aβ) production. Onjisaponin B reduces radiation-induced cell apoptosis. Onjisaponin B has anti-oxidant and anti-inflammatory activities. Onjisaponin B can be used for neurological disease and radiation injury study, and its metabolite tenuifolin (TF) can enter the brain through the BBB .
|
-
-
- HY-N16410
-
|
5-cis-C12-HSL
|
Natural Products
Microorganisms
Source classification
|
Bacterial
|
|
N-cis-Dodec-5(Z)-enoyl-L-homoserine lactone (5-cis-C12-HSL) (Compound 2) is an acylated homoserine lactone. N-cis-Dodec-5(Z)-enoyl-L-homoserine lactone can be isolated for Mesorhizobium sp. N-cis-Dodec-5(Z)-enoyl-L-homoserine lactone restores protease and pyoverdin production of an AHL-deficient Pseudomonas aeruginosa PAO1 lasI rhlI double mutant. N-cis-Dodec-5(Z)-enoyl-L-homoserine lactone has no significant antibacterial activity and cytotoxicity against tumor cells .
|
-
-
- HY-W744699
-
|
(+)-Larixol
|
Larix decidua Miller
Natural Products
Pinaceae
Source classification
Plants
|
Src
ERK
Akt
|
|
Larixol is an fMLP inhibitor and also inhibits Src kinase, ERK1/2, p38 and AKT phosphorylation signals in immune regulation. Larixol can interfere with the interaction between the βγ subunit of the fMLP receptor Gi protein and its downstream molecules, thereby inhibiting fMLP-induced respiratory burst. Larixol inhibits fMLP (0.1 μM)-induced superoxide anion production (IC50: 1.98 μM), cathepsin G release (IC50: 2.76 μM), and chemotaxis. Larixol improves neutrophil hyperactivation and reduces inflammation or tissue damage. A series of Larixol derivatives were found to have inhibitory effects on FSGS-related TRPC6 functional mutants .
|
-
-
- HY-113137
-
-
| Cat. No. |
Compare |
Product Name |
Species |
Source |
Compare Products
|
| Products |
|
| Cat. No. |
|
| Species |
|
| Source |
|
| Tag |
|
| Accession |
|
| Gene ID |
|
| Molecular Weight |
|
| Purity |
|
| Endotoxin Level |
|
| Biological Activity |
|
| Appearance |
|
| Formulation |
|
| Storage & Stability |
|
| Shipping |
|
| Free Sample |
Yes
No
|
| Size |
* This product has been "discontinued".
Optimized version of product available:
|
| Cat. No. |
Product Name |
Chemical Structure |
-
- HY-16379S
-
|
|
|
Pacritinib-d8 (SB1518-d8) is the deuterium labeled Pacritinib (HY-16379). Pacritinib (SB1518) is a potent inhibitor of both wild-type JAK2 (IC50=23 nM) and JAK2 V617F mutant (IC50=19 nM). Pacritinib also inhibits FLT3 (IC50=22 nM) and its mutant FLT3 D835Y (IC50=6 nM).
|
-
-
- HY-176354S
-
|
|
|
KRAS 2B G12V mutant, Arg- 13C36, 15N4, Lys- 13C6, 15N2 is the 13C- and 15N-labeled KRAS 2B G12V mutant.
|
-
-
- HY-176358S
-
|
|
|
KRAS 2B G12C mutant, Arg- 13C36, 15N4, Lys- 13C6, 15N2 is the 13C- and 15N-labeled KRAS 2B G12C mutant.
|
-
-
- HY-176342S
-
|
|
|
KRAS 2B Q61H mutant, Arg- 13C36, 15N4, Lys- 13C6, 15N2 is the 13C- and 15N-labeled KRAS 2B Q61H mutant.
|
-
-
- HY-176359S
-
|
|
|
KRAS 2B G12D mutant, Arg- 13C36, 15N4, Lys- 13C6, 15N2 is the 13C- and 15N-labeled KRAS 2B G12D mutant.
|
-
-
- HY-176353S
-
|
|
|
KRAS 2B G13C mutant, Arg- 13C36, 15N4, Lys- 13C6, 15N2 is the 13C- and 15N-labeled KRAS 2B G13C mutant.
|
-
-
- HY-176357S
-
|
|
|
KRAS 2B G12R mutant, Arg- 13C36, 15N4, Lys- 13C6, 15N2 is the 13C- and 15N-labeled KRAS 2B G12R mutant.
|
-
-
- HY-176341S
-
|
|
|
KRAS 2B G13D mutant, Arg-Arg- 13C36, 15N4, Lys- 13C6, 15N2 is the 13C- and 15N-labeled KRAS 2B G13D mutant.
|
-
-
- HY-14588S2
-
|
|
|
Lopinavir-d7 is deuterated labeled Lopinavir (HY-14588). Lopinavir (ABT-378) is a highly potent, selective peptidomimetic inhibitor of the HIV-1 protease, with Kis of 1.3 to 3.6 pM for wild-type and mutant HIV protease. Lopinavir acts by arresting maturation of HIV-1 thereby blocking its infectivity . Lopinavir is also a SARS-CoV 3CL pro inhibitor with an IC50 of 14.2 μM .
|
-
-
- HY-179219S
-
|
|
|
RTx-303 is an orally active, selective DNA polymerase θ (Polθ) inhibitor (IC50 = 5.1 nM). RTx-303 exhibits significantly high cellular potency and strongly potentiates PARPi in BRCA1/2 mutant cells and patient-derived xenograft models. RTx-303 can be used for the study of BRCA2-mutated breast cancer .
|
-
-
- HY-W740650
-
|
|
|
Migalastat hydrochloride- 15N (GR181413A- 15N) is the 15N-labeled Migalastat hydrochloride (HY-14929A). Migalastat (GR181413A free base) hydrochloride is an orally active α-galactosidase A molecular chaperone, with an IC50 value of 0.04 μM for human α-Gal A. Migalastat binds to the active site of certain unstable mutant forms of α-galactosidase A, facilitating their transport to the lysosome. After dissociation in the acidic environment, Migalastat enables the mutant α-galactosidase A to exhibit biological activity .
|
-
-
- HY-W728096
-
|
|
|
Migalastat-d5 (GR181413A-d5 (free base)) is deuterium labeled Migalastat. Migalastat (GR181413A free base) is an orally active α-galactosidase A molecular chaperone, with an IC50 value of 0.04 μM for human α-Gal A. Migalastat binds to the active site of certain unstable mutant forms of α-galactosidase A, facilitating their transport to the lysosome. After dissociation in the acidic environment, Migalastat enables the mutant α-galactosidase A to exhibit biological activity .
|
-
-
- HY-B0282S
-
|
|
|
Acetylcholine-d4 (chloride) is the deuterium labeled Acetylcholine chloride. Acetylcholine chloride (ACh chloride), a neurotransmitter, is a potent cholinergic agonist. Acetylcholine chloride is a modulator of the activity of dopaminergic (DAergic) neurons through the stimulation of nicotinic acetylcholine receptors (nAChRs) . Acetylcholine chloride inhibits p53 mutant peptide aggregation in vitro .
|
-
-
- HY-B0282S1
-
|
|
|
Acetylcholine-d9 (chloride) is the deuterium labeled Acetylcholine chloride. Acetylcholine chloride (ACh chloride), a neurotransmitter, is a potent cholinergic agonist. Acetylcholine chloride is a modulator of the activity of dopaminergic (DAergic) neurons through the stimulation of nicotinic acetylcholine receptors (nAChRs) . Acetylcholine chloride inhibits p53 mutant peptide aggregation in vitro .
|
-
-
- HY-153951S
-
|
|
|
BTK-IN-26 (compound 18) is a potent inhibitor of Bruton's tyrosine kinase (BTK) and its C481 mutant, with IC50 values of 0.7 and 0.8 nM for BTK and BTK C481S, respectively. BTK-IN-26 can be used for cancer and autoimmune diseases research .
|
-
-
- HY-15772S1
-
|
|
|
Osimertinib- 13C,d3 is the deuterium and 13C labeled Osimertinib. Osimertinib (AZD9291) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M, respectively.
|
-
-
- HY-18690S
-
|
|
|
Enasidenib-d6 (AG-221-d6) is the deuterium labeled Enasidenib (HY-18690) . Enasidenib is an oral, potent, reversible, selective inhibitor of the IDH2 mutant enzymes, with IC50s of 100 and 400 nM against IDH2 R140Q and IDH2 R172K, respectively .
|
-
-
- HY-15772S
-
|
|
|
Osimertinib-d6 is a deuterium labeled osimertinib. Osimertinib is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
|
-
-
- HY-W653989
-
|
|
|
Desonide- 13C3 is the 13C labeled isotope of Desonide (HY-B0248). Desonide is a non-fluorinated corticosteroid anti-inflammatory agent that acts on the glucocorticoid receptor. Desonide can also specifically bind to the mutant huntingtin protein (mHTT), reducing the level and toxicity of mHTT. Desonide can be used in the research of Huntington's disease and inflammatory diseases such as atopic dermatitis .
|
-
-
- HY-10574S
-
|
|
|
Rilpivirine-d6 is the deuterium labeled Rilpivirine. Rilpivirine (R278474) is a potent and specific diarylpyrimidine (DAPY) non-nucleoside reverse transcriptase inhibitor (NNRTI). Rilpivirine has high antiviral activity against wild-type HIV (EC50=0.4 nM) and mutant viruses (EC50=0.1-2.0 nM). Rilpivirine has a high genetic barrier to resistance development of HIV .
|
-
-
- HY-10574S1
-
|
|
|
Rilpivirine- 13C6 (R278474- 13C6) is 13C labeled Rilpivirine. Rilpivirine (R278474) is a potent and specific diarylpyrimidine (DAPY) non-nucleoside reverse transcriptase inhibitor (NNRTI). Rilpivirine has high antiviral activity against wild-type HIV (EC50=0.4 nM) and mutant viruses (EC50=0.1-2.0 nM). Rilpivirine has a high genetic barrier to resistance development of HIV .
|
-
-
- HY-124089S
-
|
|
|
Eicosapentaenoyl ethanolamide-d4 is the deuterium labeled Eicosapentaenoyl ethanolamide. Eicosapentaenoyl ethanolamide, an omega-3 fatty acid, is one of N-acylethanolamines (NAEs). Eicosapentaenoyl ethanolamide is cannabinoid CB1/CB2 receptor agonist. Eicosapentaenoyl ethanolamide acts as a metabolic signal. Eicosapentaenoyl ethanolamide inhibits dietary restriction (DR)-induced lifespan extension in wild type animals and suppresses lifespan extension in a TOR pathway mutant .
|
-
-
- HY-16767S1
-
|
|
|
Doravirine-13C,d3 (MK-1439-13C,d3) is the deuterium labeled Doravirine (HY-16767). Doravirine (MK-1439) is a highly specific HIV-1 nonnucleoside reverse transcriptase inhibitor with IC50s of 4.5 nM, 5.5 nM and 6.1 nM against the wild type and K103N and Y181C reverse transcriptase mutants, respectively .
|
-
-
- HY-114436S
-
|
|
|
MRTX-1257-d6 is the deuterium labeled MRTX-1257 (HY-114436). MRTX-1257 is a selective, irreversible, covalent and orally active KRAS G12C inhibitor, with an IC50 of 900 pM for KRAS dependent ERK phosphorylation in H358 cells .
|
-
-
- HY-14588S1
-
|
|
|
Lopinavir-d8 (ABT-378-d8) is the deuterium labeled Lopinavir. Lopinavir (ABT-378) is a highly potent, selective peptidomimetic inhibitor of the HIV-1 protease, with Kis of 1.3 to 3.6 pM for wild-type and mutant HIV protease. Lopinavir acts by arresting maturation of HIV-1 thereby blocking its infectivity . Lopinavir is also a SARS-CoV 3CLpro inhibitor with an IC50 of 14.2 μM .
|
-
-
- HY-151903S
-
|
|
|
FGFR2/3-IN-1 is a potent and selective FGFR2 and FGFR3 (FGFR) inhibitor with IC50s of 1 nM and 0.5 nM, respectively. FGFR2/3-IN-1 displays >40-fold selectivity over FGFR1/FGFR4 and other kinome. FGFR2/3-IN-1 also inhibits FGFR3 V555L and V555M mutants with IC50s of 2.7 nM and 6.1 nM, respectively .
|
-
-
- HY-15164AS
-
|
|
|
Icotinib-d4 is the deuterium-labeled Icotinib (HY-15164A). Icotinib-d4 (BPI-2009) is a potent and specific EGFR inhibitor with an IC50 of 5 nM; also inhibits mutant EGFR L858R, EGFR L858R/T790M, EGFR T790M and EGFR L861Q. Icotinib-d4 is a click chemistry reagent, itcontains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
-
- HY-W754911
-
|
|
|
Avapritinib-d3 (BLU-285-d3) is deuterium labeled Avapritinib. Avapritinib (BLU-285) is a highly potent, selective, and orally active KIT and PDGFRA activation loop mutant kinases inhibitor with IC50s of 0.27 and 0.24 nM for KIT D816V and PDGFRA D842V, respectively. Avapritinib (BLU-285) binds the active conformation of the kinase and shows antitumor activity. Avapritinib (BLU-285) attenuates the transport function of both ABCB1 and ABCG2 .
|
-
-
- HY-14588S
-
|
|
|
(rel)-Lopinavir-d8 ((rel)-ABT-378-d8) is the deuterium labeled Lopinavir (HY-14588) . Lopinavir (ABT-378) is a highly potent, selective peptidomimetic inhibitor of the HIV-1 protease, with Kis of 1.3 to 3.6 pM for wild-type and mutant HIV protease. Lopinavir acts by arresting maturation of HIV-1 thereby blocking its infectivity . Lopinavir is also a SARS-CoV 3CL pro inhibitor with an IC50 of 14.2 μM .
|
-
-
- HY-13238S2
-
|
|
|
Dolutegravir-d5 is deuterium labeled Dolutegravir. Dolutegravir (S/GSK1349572) is a highly potent and orally bioavailable HIV integrase strand transfer inhibitor with an IC50 of 2.7 nM for HIV-1 integrase-catalyzed strand transfer. Dolutegravir (S/GSK1349572) inhibits HIV-1 viral replication with an IC50 of 0.51 nM in peripheral blood mononuclear cells. Dolutegravir retains a high potency against the HIV-1 Y143R, N155H, and G140S/Q148H mutants (EC50=3.6-5.8 nM) .
|
-
-
- HY-13238S1
-
|
|
|
Dolutegravir-d3 is the deuterium labeled Dolutegravir. Dolutegravir (S/GSK1349572) is a highly potent and orally bioavailable HIV integrase strand transfer inhibitor with an IC50 of 2.7 nM for HIV-1 integrase-catalyzed strand transfer. Dolutegravir (S/GSK1349572) inhibits HIV-1 viral replication with an IC50 of 0.51 nM in peripheral blood mononuclear cells. Dolutegravir retains a high potency against the HIV-1 Y143R, N155H, and G140S/Q148H mutants (EC50=3.6-5.8 nM) .
|
-
| Cat. No. |
Compare |
Product Name |
Application |
Reactivity |
Compare Products
|
| Products |
|
| Cat. No. |
|
| Host |
|
| Reactivity |
|
| Application |
|
Dilution
Ratio |
|
| Molecular Weight |
|
| Conjugation |
|
| Clonality |
|
| Immunogen |
|
| Appearance |
|
| Isotype |
|
| Gene ID |
|
| SwissProt ID |
|
| Purity |
|
| Formulation |
|
| Free Sample |
Yes
No
|
| Size |
* This product has been "discontinued".
Optimized version of product available:
|
| Cat. No. |
Product Name |
|
Classification |
-
- HY-159493
-
|
|
|
Azide
|
|
BI2536-PEG2-Halo is a bifunctional molecule containing a ligand for Halo tag and a Polo-like kinase 1 (PLK1) inhibitor BI-2536 (HY-50698). BI2536-PEG2-Halo inhibits the proliferation of 293T cells with Halo-p53R273H(FL)-mCherry tag (IC50=23 nM), exhibits selective toxicity against p53 mutant cancer cells .
|
-
- HY-134813
-
MRTX1133
Maximum Cited Publications
32 Publications Verification
|
|
Alkynes
|
|
MRTX1133 is a noncovalent, potent, and selective alkyne-based KRAS G12D inhibitor. MRTX1133 optimally fills the switch II pocket and extends three substituents to favorably interact with the protein, resulting in an estimated KD against KRAS G12D of 0.2 pM. MRTX1133 prevents SOS1-catalyzed nucleotide exchange and/or formation of the KRAS G12D/GTP/RAF1 complex, thereby inhibiting mutant KRAS-dependent signal transduction. MRTX1133 selectively inhibits KRAS G12D mutant, but not KRAS wild-type, tumor cells. MRTX1133 has single digit nanomolar activity in cellular assays and marked in vivo efficacy in tumor models harboring KRAS G12D mutations .
|
-
- HY-15164A
-
|
BPI-2009
|
|
Alkynes
|
|
Icotinib (BPI-2009) is a potent and specific EGFR inhibitor with an IC50 of 5 nM; also inhibits mutant EGFR L858R, EGFR L858R/T790M, EGFR T790M and EGFR L861Q. Icotinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
- HY-15164
-
|
BPI-2009H
|
|
Alkynes
|
|
Icotinib Hydrochloride (BPI-2009) is a potent and specific EGFR inhibitor with an IC50 of 5 nM; also inhibits mutant EGFR L858R, EGFR L858R/T790M, EGFR T790M and EGFR L861Q. Icotinib (Hydrochloride) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
- HY-149880
-
|
|
|
Alkynes
|
|
c-Met-IN-18 is ATP competitive type-III c-MET inhibitor of WT and D1228V mutant c-MET. c-Met-IN-18 has inhibitory for WT/D1228V with an IC50 value of 0.013/0.20 e.c-Met-IN-18 can be used for the research of c-MET driven cancers . c-Met-IN-18 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
- HY-176870
-
|
|
|
Alkynes
|
|
KRAS-IN-42 (Compound Z1063) is a covalent KRAS G12D mutants inhibitor. KRAS-IN-42 is promising for research of KRAS G12D-mutant cancers (e.g., non-small cell lung cancer, colorectal cancer) .
|
-
- HY-167690
-
|
|
|
Alkynes
|
|
MK-6186 is a novel non-nucleoside reverse transcriptase inhibitor with sub-nanomolar activity against wild-type viruses and the two most common NNRTI-resistant RT mutants (K103N and Y181C). MK-6186 exhibits excellent antiviral activity against K103N and Y181C mutant viruses. When MK-6186 targets 12 common NNRTI-associated mutant viruses, only two relatively rare mutants (Y188L and V106I/Y188L) show high resistance, with FC values exceeding 100, while the FC values of the remaining viruses are all below 10. In addition, when MK-6186 faces 96 clinical virus isolates carrying NNRTI-resistant mutations, most (70%) viruses show more than 10-fold resistance to efavirenz (EFV), while only 29% of mutant viruses show more than 10-fold resistance to MK-6186 .
|
-
- HY-159476
-
|
|
|
Azide
|
|
KRAS inhibitor-26 (compound 194a) is a potent KRAS G12V inhibitor (IC50: ≤100 nM). KRAS inhibitor-26 can be used for cancer research .
|
-
- HY-179300A
-
|
|
|
Alkynes
|
|
KRAS-IN-48 (Compound 1-01) is a KRAS mutant inhibitor, with Kd values of 2.58 nM and 5.49 μM for KRAS-G12D and KRAS-G12V, respectively. KRAS-IN-48 can be used in the research of cancer .
|
-
- HY-178755
-
|
|
|
Alkynes
|
|
DEL1187-126-5-80 is a JAK3 inhibitor with IC50s of 52 and 7.9 nM against JAK3 WT in 0 and 60 min. DEL1187-126-5-80 has no activity at all towards the C909S mutant, JAK1, JAK2, and TYK2. DEL1187-126-5-80 can be used for the study of immune-mediated diseases .
|
-
- HY-131033
-
|
|
|
Azide
|
|
L-Azidonorleucine hydrochloride, an unnatural amino acid, is A Methionine surrogate. L-Azidonorleucine hydrochloride can be used to label mammalian cell proteins and identify a diverse set of methionyl-tRNA synthetase (MetRS) mutants . L-Azidonorleucine (hydrochloride) is a click chemistry reagent, it contains an Azide group and can undergo copper-catalyzed azide-alkyne cycloaddition reaction (CuAAc) with molecules containing Alkyne groups. It can also undergo strain-promoted alkyne-azide cycloaddition (SPAAC) reactions with molecules containing DBCO or BCN groups.
|
-
- HY-104066A
-
|
Xiliertinib tartrate; HMPL-309 tartrate
|
|
Alkynes
|
|
Theliatinib (Xiliertinib) tartrate is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib (tartrate) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
- HY-172140
-
|
|
|
Alkynes
|
|
Ebio3 is a selective potassium channel (KCNQ2) inhibitor with an IC50 of 1.2 nM. Ebio3 binds to the KCNQ2 channel through its hydrophobic tail, causing the S6 helix to move inward, which leads to the closure of the inner gate. The inhibitory effect of Ebio3 is also effective in pathogenic mutants of KCNQ2 (such as R75C and I238L), where it can inhibit outward currents by more than 80%. Ebio3 is expected to be used in the research of neurological diseases such as epilepsy .
|
-
- HY-104066
-
|
Xiliertinib; HMPL-309
|
|
Alkynes
|
|
Theliatinib (Xiliertinib) is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
- HY-128522
-
|
|
|
Alkynes
|
|
ARS-1323-alkyne is a covalent inhibitor probe that covalently binds to the Switch-II pocket (S-IIP) of the KRAS G12C mutant protein. ARS-1323-alkyne visualizes the covalent modification of KRAS G12C and quantitatively measures the binding efficiency of the inhibitor to the target. ARS-1323-alkyne can be used to validate the target occupancy of KRAS G12C inhibitors and the synergistic mechanism of combination therapy, providing tool support for the development of combination treatment strategies .
|
-
- HY-152233
-
|
|
|
Azide
|
|
Reverse transcriptase-IN-4 (compound F10) is a potent and selective non-nucleoside reverse transcriptase (NNRT) inhibitor with an EC50 value of 0.053 μM for wild-type HIV-1 and an EC50 value of 0.26 μM for HIV-1 mutant E138K . Reverse transcriptase-IN-4 is a click chemistry reagent, it contains an Azide group and can undergo copper-catalyzed azide-alkyne cycloaddition reaction (CuAAc) with molecules containing Alkyne groups. It can also undergo strain-promoted alkyne-azide cycloaddition (SPAAC) reactions with molecules containing DBCO or BCN groups.
|
| Cat. No. |
Product Name |
|
Classification |
-
- HY-111627
-
|
|
|
Nucleoside Analogs
Guanosine
|
|
5-Aza-7-deazaguanine is a substrate for wild-type (WT) E. coli purine nucleoside phosphorylase and its Ser90Ala mutant in the synthesis of base-modified nucleosides .
|
-
- HY-132609
-
|
|
|
siRNAs
siRNA drugs
|
|
Patisiran sodium is a double-stranded small interfering RNA that targets a sequence within the transthyretin (TTR) messenger RNA. Patisiran sodium specifically inhibits hepatic synthesis of mutant and wild-type TTR. Patisiran sodium can be used for the research of hereditary TTR amyloidosis .
|
-
- HY-145411
-
|
|
|
Pegylated Lipids
|
|
PEG2000-C-DMG, a pegylated lipid, can be used for the preparation of Onpattro. Onpattro, a hepatically directed investigational RNAi therapeutic agent, harnesses this process to reduce the production of mutant and wild-type transthyretin by targeting the 3′ untranslated region of transthyretin mRNA .
|
-
- HY-153609
-
|
|
|
siRNAs
siRNA drugs
|
|
AS-Patisiran sodium is an antisense strand of Patisiran. Patisiran is a double-stranded small interfering RNA that targets a sequence within the transthyretin (TTR) messenger RNA. Patisiran specifically inhibits hepatic synthesis of mutant and wild-type TTR. Patisiran can be used for the research of hereditary TTR amyloidosis .
|
-
- HY-171499
-
|
EL625
|
|
Antisense Oligonucleotides
|
|
Cenersen (EL625) is an oligonucleotide targeting TP53. Cenersen can eliminate the activity of TP53 gain-of-function mutants and increase the sensitivity of lymphoma cells to cytotoxic chemotherapy in vitro. Cenersen can be used for the study of chronic lymphocytic leukemia (CLL) .
|
-
- HY-160044
-
|
|
|
Aptamers
|
|
Apt-M13 aptamer sodium is a mutant 33 nt aptamer based on the original aptamer for dexamethasone (HY-14648). Apt-M13 aptamer sodium can be combined with other nanomaterials for rapid detection of dexamethasone (HY-14648) .
|
-
- HY-177453
-
|
DYNE-101 oligomer
|
|
Antisense Oligonucleotides
|
|
Basivarsen (DYNE-101 oligomer) is composed of an antisense oligonucleotides and a linker (HY-177454). It is designed to target mutant nuclear myotonic dystrophy protein kinase (DMPK) RNA for RHase H-mediated degradation to correct splicing. It is used for the study of myotonic dystrophy type 1 (DM1).
|
Your information is safe with us. * Required Fields.
Inquiry Information
- Product Name:
- Cat. No.:
- Quantity:
- MCE Japan Authorized Agent:








































































































































































































































































![PIP-C-3-Azaspiro[5.5]undecane-boc](http://file.medchemexpress.eu/product_pic/hy-161656.gif)























































































































![[Ala28]-β Amyloid(25-35)](http://file.medchemexpress.eu/product_pic/hy-p5968.gif)









![[Ala28]-β Amyloid(25-35) TFA](http://file.medchemexpress.eu/product_pic/hy-p5968a.gif)

































































































































































































































































































































































































































































































































































































































